https://doi.org/10.22319/rmcp.v16i1.6616
Nota de investigación
Comparación de métodos de aprendizaje automático para predicción de valores de cría genómicos en características de crecimiento en bovinos Suizo Europeo
José Luis Velez Labrada a
Paulino Pérez Rodríguez b*
Mohammad Ali Nilforooshan c
Agustín Ruíz Flores a*
a Universidad Autónoma Chapingo. Posgrado en Producción Animal. Carretera Federal México-Texcoco Km 38.5, 56230, Texcoco, Estado de México, México.
b Colegio de Postgraduados. Campus Montecillo. Socioeconomía Estadística e Informática. Texcoco, Estado de México, México.
c Livestock Improvement Corporation, Hamilton, New Zealand.
*Autores de correspondencia: aruizf@chapingo.mx y perpdgo@colpos.mx.
Resumen:
Los algoritmos de Aprendizaje Automático (AA) han demostrado ventaja al abordar desafíos asociados con la cantidad y la complejidad de la información, permiten descubrir patrones, realizar análisis eficientes y servir como herramienta para la toma de decisiones. Este estudio, tuvo como objetivo comparar cuatro métodos de AA: redes neuronales artificiales (RN), árboles de regresión (AR), bosques aleatorios (BA) y máquina de soporte vectorial (SVM) para predecir el valor genómico en bovinos Suizo Europeo utilizando registros fenotípicos de pesos al nacimiento (PN), destete (PD) y al año (PA), así como información genómica. Los resultados indican que la capacidad predictiva de los modelos varía según la característica y la cantidad de información disponible. En general, RN, BA y SVM mostraron un desempeño similar, mientras que AR tuvo un desempeño inferior. La metodología SVM destacó como la herramienta con mayor potencial, obteniendo los valores más altos de correlación Pearson entre fenotipos corregidos y valores genéticos predichos para PD. A pesar de un mayor costo computacional, RN tuvo un desempeño razonable, especialmente para PN y PA. La selección del modelo final depende de las necesidades particulares de la aplicación, así como de factores prácticos como la disponibilidad de datos, recursos computacionales y la interpretabilidad; pero en general, RN y SVM surgieron como opciones sólidas en varias categorías.
Palabras clave: Árboles de regresión, Bosques aleatorios, Redes neuronales, Capacidad predictiva.
Recibido: 08/01/2024
Aceptado: 28/11/2024
La genómica ha evolucionado en años recientes gracias a los avances en la tecnología de secuenciación de ADN. Estos avances han permitido la generación de grandes cantidades de datos a una velocidad sin precedentes. Sin embargo, por la complejidad inherente a los datos genómicos, así como su dimensionalidad, plantean obstáculos importantes(1). La diversidad de la información genómica, que abarca desde secuencias de ADN hasta datos fenotípicos asociados, añade una capa adicional de complejidad. Además, la variabilidad en la calidad y la estructura de los datos genómicos puede dificultar la extracción de conocimientos útiles y significativos. En este contexto, los métodos de aprendizaje automático (AA) emergen como herramientas valiosas para abordar estos desafíos, estas metodologías ofrecen la capacidad de procesar y analizar grandes volúmenes de datos de manera eficiente y precisa(2). Su capacidad para identificar patrones complejos y relaciones no lineales en datos genómicos y fenotípicos los convierte en una herramienta poderosa para la extracción de conocimientos(2,3).
La aplicación de técnicas de AA permite abordar tareas como la identificación de genes relevantes para características específicas, predicción de funciones génicas, detección de variantes genéticas asociadas con características particulares y clasificación de especies con base en información genómica(4,5,6). Recientemente, el AA se ha vuelto atractivo en la predicción genómica por su capacidad para manejar grandes volúmenes de datos, su flexibilidad en el modelar relaciones no lineales, mejorar la precisión predictiva y las continuas innovaciones en algoritmos y técnicas, pero es necesario investigar cómo se compara en la predicción de valores genéticos con los métodos GBLUP convencionales(7). Por lo que la combinación de datos genómicos con algoritmos de AA permitiría la creación de modelos predictivos y descriptivos confiables, que a su vez tendría implicaciones en la cría selectiva, conservación de especies y la comprensión de la evolución(8,9).
Entre los métodos de AA más utilizados están las redes neuronales, máquinas de soporte vectorial, árboles de decisión, regresión lineal y métodos de agrupación(3,8-11). La diversidad de enfoques disponibles refleja la versatilidad de estos métodos en la resolución de desafíos con información genómica, como la clasificación de secuencias de ADN y la predicción de la estructura de proteínas(12). El éxito de la aplicación de estos métodos en la genómica animal depende en gran medida de la disponibilidad de información(13). Además, de elegir el método de AA óptimo, ya que se han propuesto una serie de métodos, cada uno con características propias y capacidades de predicción específicas con diferentes conjuntos de datos y características(3,7).
Por lo expuesto el presente estudio tuvo como objetivo comparar los métodos de AA: redes neuronales (RN), árboles de regresión (AR), bosques aleatorios (BA), y máquina de soporte vectorial (SVM) para predecir valores genómicos de cría utilizando registros fenotípicos de pesos al nacimiento, destete y al año, así como la información genómica de una población de bovinos Suizo Europeo en México.
La información utilizada provino de la base de datos de la Asociación Mexicana de Criadores de Ganado Suizo de Registro (AMCGSR), la cual contiene registros fenotípicos e identificación de los animales, rancho de origen o propietario, genealogía y de caracteres de importancia económica como pesos al nacimiento (PN), al destete (PD) y al año (PA). El conjunto de datos utilizado fue analizado previamente por Valerio-Hernández et al(14,15) para ajustar otros modelos, por lo que algunos resultados de los aquí obtenidos se comparan en forma directa con los de los autores mencionados. El tratamiento de la información fenotípica para PN, PD y PA se realizó el procedimiento descrito por Valerio-Hernández et al(14,15), es decir, se omitieron individuos con información faltante en edad de la madre, manejo, hato de procedencia, así como individuos no relacionados genéticamente. Los grupos contemporáneos (GC) se definieron combinando los efectos de hato, año y época de nacimiento. Para PD los GC se conformaron considerando el manejo alimenticio que se da al hato, así como ajuste a días específicos para el destete. Se descartaron GC con menos de tres individuos o con varianza cero, de acuerdo con la metodología citada(14).
La información genómica se obtuvo a través del análisis de muestras de pelo recolectadas de 300 animales de ranchos pertenecientes a la AMCGSR ubicados en Colima, Jalisco y Veracruz. El genotipado lo realizó la empresa GeneSeek (Lincoln, NE, USA), mediante el chip Genomic Profile Bovine LDv.4, el cual se ha utilizado para genotipar diversas razas Bos indicus y Bos taurus. El genotipado de 150 animales se hizo con un chip de 30,000, otros 150 animales se genotiparon con 50,000 marcadores SNP (Single Nucleotide Polymorphism). Se realizó una selección de marcadores SNP que estuvieran presentes en ambos chips, lo que resultó en un conjunto de 12,835 SNP en común.
La recodificación y control de calidad de la información genotípica realizada por Valerio-Hernández et al(15), se basó en lo realizado por Jarquín et al(16) recodificando para efectos genéticos aditivos como AA=0, AB=1 y BB=2. Para la imputación de genotipos faltantes en el presente estudio, se utilizó el software FImpute(17) (versión 2.2), para lo cual se obtuvo: 1) el mapa de los marcadores (marcador, cromosoma, posición en pares de base), eliminando marcadores duplicados o marcadores con posiciones desconocidas, 2) el pedigrí de los individuos y su correspondiente sexo. Se eliminaron marcadores monomórficos y aquellos con una frecuencia del alelo menor (MAF) más pequeña que 0.04. Se obtuvieron 9,008 marcadores que se utilizaron para la construcción de la matriz de relaciones genómicas G; en el Cuadro 1, se muestra el número de animales que se incorporaron en el estudio por cada característica después del filtrado.
Cuadro 1: Número de animales genotipados y fenotipados de una población de bovinos Suizo Europeo para tres características de crecimiento
PN= peso al nacimiento, PD= peso al destete, PA= peso al año. G2 Animales con fenotipos e información genómica.
La matriz de relaciones genómicas G se obtuvo de acuerdo con la metodología descrita por Pérez-Rodríguez et al(18), G=WWt/p, donde W es la matriz de marcadores centrada y estandarizada y p el número total de marcadores. Adicionalmente se calculó la matriz de relaciones H que combina información de la matriz G con la información de la matriz de relaciones genéticas aditivas A, obtenida para los individuos con pedigrí.
Modelos mixtos lineales (modelos base). Con el objetivo de comparar los resultados del poder predictivo para valores de cría para PN, PD y PA para modelos mixtos lineales vs modelos de aprendizaje automático se consideran la secuencia de modelos y de resultados descritos por Valerio-Hernández et al(15). Con la finalidad de presentar toda la información pertinente se describe a continuación en modelo mixto lineal utilizado por el mencionado autor, el cual está dado por:
y=Xb+Z1c+Z2a+e, … (1)
donde y es el vector de fenotipos, X es la matriz de incidencias para efectos fijos, que para este estudio son: sexo del animal, edad de la madre de cada animal y el grupo contemporáneo ya descritas, b es el vector de efectos fijos, Z1 es una matriz de incidencias que conecta los fenotipos con los grupos contemporáneos, cuyos efectos se suponen aleatorios y representan la variabilidad en los fenotipos que se debe a diferencias entre grupos de individuos que están sujetos a las mismas condiciones ambientales y de manejo, c~NM(0,gc2I), con NM que denota la distribución normal multivariada, con media 0 y gc2 parámetro de varianza asociado, I la matriz identidad, Z2 es una matriz es una matriz de incidencias que conecta los fenotipos con efectos genéticos aditivos los cuales se suponen como efectos aleatorios, a∼NM(0,a2K), con K∈{A,G,H}, e~NM(0,e2I) representa el vector de errores aleatorios, donde e2 denota la variabilidad asociada con los mismos. Dependiendo de los datos utilizados, el modelo (1) da lugar a tres modelos distintos que se denotan como sigue: 1) BLUP, K=A, 2) GBLUP, K=G y 3) ssGBLUP (GBLUP en un solo paso) K=H. Los modelos mixtos lineales descritos previamente fueron ajustados por Valerio-Hernández et al(15) utilizando el paquete estadístico BGLR(19).
Modelos de aprendizaje automático. Las variables de entrada para los algoritmos de AA que se utilizaron fueron la matriz de relaciones genéticas que combina la información genómica e información de pedigrí denominada H, así como los efectos de edad de la madre para cada animal, variables indicadoras de sexo, grupo contemporáneo descritas previamente. Para poder incluir la información de la matriz H en los modelos de aprendizaje se realizó la descomposición espectral de la misma, es decir, H=ΓΛt a partir de la cual se obtuvo X=12 (componentes principales) mismos que se utilizaron como covariables (variables explicatorias) en los modelos, esta estrategia computacional y otras relacionadas han sido utilizadas por otros autores anteriormente(20,21).
Red neuronal artificial. Las redes neuronales (RN) son modelos que en sus inicios pretendían emular el funcionamiento del sistema nervioso, donde a través de operadores matemáticos procesan información de entrada, generando valores de salida o el resultado final(3,22). Las variables de entrada afectan el desempeño del modelo, puede generar sobreajuste si la cantidad de información es grande por lo que es importante optimizar dichas variables(23). Una de las ventajas de las redes neuronales es su capacidad de aprender patrones no lineales(3). El modelo de una RN con una capa de entrada con p predictores, una capa oculta con S neuronas y una capa de salida con una respuesta continua puede expresarse como:
yi= 0+k=1swkg(0k+j=1pj(k)xij)+ei,
donde ei~NIID(0,e2), con NIID denotando variables aleatorias normales, independientes e idénticamente distribuidas; k=1,…,S (neuronas); j=1,…,p (predictores); i=1,…,n (observaciones); y g(⋅) es la función de activación, de acuerdo con Bai et al(24) y Gianola et al(20), donde, yi es la variable respuesta para el i-ésimo individuo, para este caso los pesos de características de crecimiento en bovinos Suizo Europeo (PN, PD, PA) que la red predice en función de las entradas; 0 es el término de sesgo o el intercepto, éste puede representar el valor predicho cuando las entradas son igual a cero y wk son pesos asociados a cada una de las neuronas y determinan la contribución de cada una de ellas en la predicción final. La capa oculta es una capa intermedia entre la capa de entrada y la capa de salida, es donde se lleva a cabo la mayor parte del procesamiento y la extracción de características del conjunto de datos; está compuesta por un número específico de neuronas (S) es un hiperparámetro del modelo que se ajusta durante el proceso de entrenamiento. Un valor más alto de S permite que la red neuronal capture una mayor complejidad en los datos, pero también puede aumentar el riesgo de sobreajuste. La RN ajusta los parámetros ('s, w's) durante el proceso de entrenamiento para minimizar el error de predicción. La función de activación, g(⋅), mapea las entradas de la línea real al intervalo abierto acotado (–1,1), ejemplificado por Pérez-Rodríguez et al(25) donde gx=2/[1+exp -2x ]-1 se conoce como la función de activación tangente hiperbólica (tanh). Para el ajuste del modelo de red neuronal se utilizó la función “brnn”(26) incluida en el paquete del mismo nombre (versión 0.9.3) en el paquete estadístico R(27) (versión 4.3.0).
Árboles de regresión. Este modelo se basa en el planteado por Breiman et al(28), yi= j=1JyjI(xi Rj), donde yi es una variable respuesta (PN, PD y PA), yj es el valor de regresión que asociado con una “hoja”, xi es el conjunto de características de la observación, Rj es la región asociada a la “hoja j” definida por características y valores de corte en el camino desde la “raíz” hasta la “hoja”. I(⋅) es una función indicadora que toma el valor 1 si la observación i pertenece a la región Rj. El árbol busca encontrar las divisiones que minimicen el error en cada región y dividir recursivamente hasta alcanzar un criterio de parada del proceso, como la profundidad máxima del árbol o el número mínimo de casos en una hoja. El ajuste del modelo se realizó con la función “rpart”(29) incluida en la biblioteca de funciones del mismo nombre (versión 4.1.19) en el paquete estadístico R(27) (versión 4.3.0).
Bosques aleatorios. Este modelo combina múltiples AR donde las predicciones de cada uno se promedian para obtener una predicción final optimizada, yi= 1Nj=1Nyij, donde N es el número de árboles del bosque aleatorio, yi es una variable aleatoria observada (PN, PD y PA) y yij es la predicción del j-ésimo AR para la observación i. La implementación del algoritmo bosques aleatorios se realizó utilizando la función “randomForest” (30) (versión 4.7-1.1) incluida en la biblioteca de funciones del mismo nombre en el paquete estadístico R(27) (versión 4.3.0).
Máquina de soporte vectorial. El modelo de máquina de soporte vectorial (SVM), se utiliza para clasificación y regresión(31). En el contexto de regresión, dado un conjunto de datos y1,x1,…,{yn,xn}, donde yi representa el valor de la variable respuesta (continua) para el i-ésimo individuo, xi el valor de las covariables asociadas, el objetivo es obtener una función f(x) de tal forma que la distancia con y no sea más grande que para cada uno de los puntos de entrenamiento. De acuerdo con Hastie et al(32) la función de regresión se aproxima en términos de funciones base hmx,m=1,…,M como sigue:
fx=0+m=1Mmhm(x),
donde β=(β0,1,…,M)t son coeficientes que se obtienen al minimizar: Q=i=1nLyi-fxi+2m2, en la que L(⋅) se denomina función de pérdida (por ejemplo, cuadrática o valor absoluto) y es un parámetro de regularización positivo. Para cualquier selección de L(⋅), la solución tiene la forma: fx=i=1naiK(x,xi), con K(⋅,⋅) conocida como función kernel. Los kernel se consideran en el modelo como elementos fundamentales, sirven como funciones que permiten transformar los datos y generar un espacio de mayor dimensión, ayudan a modelar relaciones complejas en los datos. Los kernel más comunes son el lineal (xitxj), polinomial (xitxj+coef0)d,d=2,3,…, radial (e-γ||xi-xj||2) y sigmoide (tanh(xitxj), +coef0), donde se conoce como ancho de banda que se ajusta en el proceso de entrenamiento mediante validación cruzada, coef0 es una constante que se puede ajustar durante el proceso de entrenamiento del modelo, aunque usualmente se fija a 1. El ajuste del modelo se realizó con el paquete “e1071”(33) (versión 1.7-13), con ayuda de la función svm en el paquete estadístico R(27) (versión 4.3.0). Los códigos para ajuste de los modelos están disponibles previa solicitud al autor para correspondencia.
Validación cruzada. La validación cruzada es un método de re-muestreo de datos muy utilizado para estimar el verdadero error de predicción de los modelos y ajustar los parámetros del modelo(20,34). Por tanto, para obtener la capacidad de predicción de los modelos RN, AR, BA y SVM, y con ello hacer la comparación, se realizó la validación cruzada teniendo como referencia los procedimientos realizados por Valerio-Hernández et al(15). Estos autores dividieron los datos aleatoriamente en 80 % para el conjunto de entrenamiento y 20 % para el de validación y el proceso se repitió 100 veces. Se ajustaron los modelos de AA y se obtuvieron las correlaciones entre los valores observados vs predichos, donde los valores observados se consideraron los valores de la variable respuesta corregidos por efectos fijos y otros efectos aleatorios. Se obtuvo el coeficiente de correlación de Pearson de los fenotipos corregidos y valores genéticos predichos para cada una de las particiones y se obtuvieron los promedios para cada uno de los modelos.
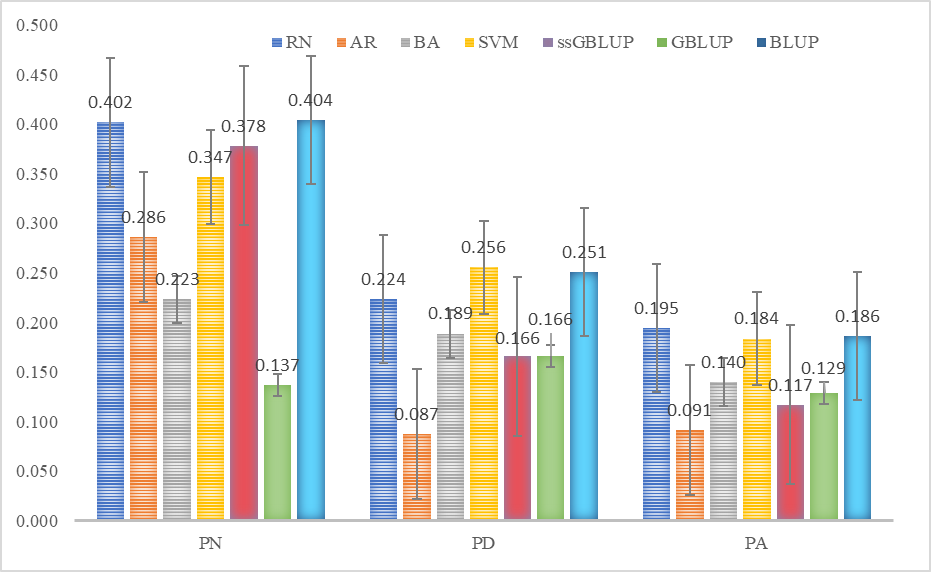
En el Cuadro 2 se muestran los promedios de las cien correlaciones de Pearson (en validación cruzada) entre valores corregidos y predichos para las características PN, PD y PA, utilizando los cuatro algoritmos de AA comparados en el estudio. Para PD, el algoritmo SVM fue con el que se obtuvieron los valores más altos para los coeficientes de correlación de Pearson entre valores corregidos y predichos en los conjuntos de validación (PD= 0.256). En este método para las tres características el mejor ajuste se obtuvo utilizando el “Kernel Radial” optimizando los hiperparámetros (gamma) y costo (PN: 0.045 y 0.05; PD y PA: 0.05 y 0.01). Con la metodología de RN Artificial se realizaron varias pruebas en relación con el número de neuronas en la capa oculta del modelo, siendo 3 neuronas para PN y 2 para PD y PA cuando se obtuvieron estimadores de los parámetros adecuados de ponderadores generando un modelo parsimonioso. Para PN y PA, este algoritmo obtuvo el mejor desempeño con 0.402 y 0.195, respectivamente.
Con la metodología BA se realizaron pruebas con diferente cantidad de “árboles” dentro de los parámetros del modelo, siendo 150 árboles para PN y PD; y 250 para PA los que obtuvieron valores óptimos de predicción; para las características PD y PA obtuvieron el tercer mejor rendimiento en cuestión de sus predicciones. En relación con la metodología AR mostró menor capacidad predictiva para las características PD y PA de esta investigación.
Con los resultados obtenidos y con la finalidad de probar la significancia de los coeficientes de correlación obtenidos se planteó el siguiente juego de hipótesis: H0:r≤0 vs H1:r>0, donde r es la media de la distribución del coeficiente de correlación de Pearson y se desea probar si la asociación es o no positiva. El juego de hipótesis planteado fue contrastado utilizando la prueba de t para 1 muestra, verificando primero el supuesto de normalidad en cada uno de los casos(35), en todos los casos se concluyó que el supuesto de normalidad es adecuado (P-valor>0.05).
Cuadro 2: Promedios de los estimadores de la correlación Pearson y desviación estándar entre fenotipos corregidos y valores genéticos predichos, para las 100 validaciones cruzadas para las tres características de crecimiento y los algoritmos comparados
PN= peso al nacimiento, PD= peso al destete, PA= peso al año; Cor = coeficiente de correlación de Pearson; DE= desviación estándar de los 100 estimadores de la correlación para particiones seleccionadas al azar.
Para determinar la capacidad predictiva de los modelos de AA, se compararon los estimadores del coeficiente de correlación Pearson entre los fenotipos corregidos y los valores genéticos predichos con los modelos comparados(36), esto realizado en los conjuntos de prueba para cada característica de la metodología validación cruzada descrita anteriormente, donde no se produjo variación en la información utilizada en los análisis en comparación con otros estudios previos. Esto garantiza la consistencia en las comparaciones realizadas y proporciona una base sólida para evaluar el rendimiento relativo de los métodos tradicionales y los algoritmos de AA. El problema de inferencia de valores genéticos y predicción de fenotipos para caracteres cuantitativos gobernados por formas complejas de interacción entre genes resulta difícil de resolver utilizando los modelos mixtos lineales utilizados de manera rutinaria(37,38), por lo que el uso de algoritmos de AA son una alternativa para modelar funciones complejas identificando relaciones no lineales entre las covariables y la variable respuesta(20). Las correlaciones entre fenotipos corregidos y valores predichos con las metodologías utilizadas permiten evaluar los algoritmos de aprendizaje automático RN, AR, BA y SVM, para las características de crecimiento en bovinos PN, PD y PA. En la Figura 1 se muestra que en general los algoritmos RN, BA y SVM mostraron un desempeño predictivo similar a las metodologías evaluadas por Valerio-Hernández et al(15), quienes trabajaron con las mismas variables. En un estudio donde se comparó la capacidad predictiva de redes neurales no lineales (RNNL) con modelos lineales, se encontró que éstas pueden ser útiles en la predicción para características complejas utilizando información genómica, situación en la que ordinariamente el número de parámetros a estimar supera el tamaño de muestra(20). Por su parte, Rodríguez-Alcántar(3) comparó algoritmos de AA utilizando diversos conjuntos de SNP generados a partir de cromosomas con alto número de QTL asociados con alta producción lechera. Este autor encontró que la precisión de la clasificación varió de 90.9 a 94.5 % con árboles de decisión, y de 79.0 a 87.3 % con redes neuronales. El autor concluye que tanto el método de redes neuronales para clasificación binaria, como los árboles de decisión son herramientas eficientes para la identificación temprana de vacas lecheras altas productoras.
Figura 1: Comparación de los coeficientes de correlación (promedio de las 100 validaciones) de los fenotipos corregidos y valores genéticos predichos
Valores genéticos obtenidos con los métodos de aprendizaje automático, redes neuronales artificiales (RN), arboles de regresión (AR), bosques aleatorios (BA) y máquina soporte vectorial (SVM) con las metodologías realizadas por Valerio-Hernández et al(15), mejor predictor lineal insesgado (BLUP), BLUP genómico (GBLUP) y GBLUP de un solo paso (ssGBLUP) para las características peso al nacer (PN), peso al destete (PD) y peso al año (PA) de una población de bovinos Suizo Europeo.
Los resultados indican que el desempeño de los modelos varía según la característica y la cantidad de información(20), entre otros factores. Lo anterior sugiere que pueden obtenerse mejores resultados con estos modelos al incluir más información de variables y covariables para ajustar el modelo en entrenamiento(39,40), pese a las bajas correlaciones y grandes varianzas de las predicciones, estas pueden atribuirse a una serie de factores genéticos y metodológicos. En consonancia con los hallazgos de Cuyabano et al(41), es importante considerar las diferencias genéticas entre las poblaciones de referencia y las poblaciones objetivo al calcular la precisión de las predicciones. Además, sugiere que existe un límite teórico superior para la precisión de estas predicciones, que está determinado por la raíz cuadrada de la heredabilidad. Zhang et al(42) mencionan que varios factores pueden influir en la precisión de las predicciones de valores de cría genómicos; la heredabilidad (empleando el modelo descrito como BLUP, Valerio-Hernández et al(15) reporta 0.260 para PN; 0.223 para PD y 0.231 para PA), la densidad de marcadores genéticos, la frecuencia del alelo menor (MAF por sus siglas en inglés) utilizado durante el proceso de depuración de datos y el modelo estadístico utilizado son solo algunos factores que pueden afectar la precisión. Esto plantea desafíos significativos en la predicción de rasgos complejos.
Las metodologías de SVM, RN y BA mostraron un desempeño similar en términos de los coeficientes de correlación de Pearson de los fenotipos corregidos y los valores predichos para las tres características de crecimiento utilizadas; comparando los resultados de éstas con valores obtenidos por Valerio-Hernández et al(15) utilizando metodologías tradicionales BLUP, GBLUP y ssGBLUP. El costo computacional de RN fue mayor que el de los otros tres algoritmos comparados, se determinó, midiendo el tiempo de ejecución necesario para entrenar y validar cada uno de los algoritmos en los conjuntos de datos de entrenamiento y prueba, registrando el tiempo transcurrido desde el inicio del entrenamiento hasta la finalización del proceso de validación; este resultado es similar al que reportaron Zhao et al(43) quienes mencionan que el ajuste de la RN es más complicado y requiere más tiempo. El algoritmo SVM destacó como una herramienta prometedora para la predicción utilizando información genómica, considerando la cantidad de información y los parámetros utilizados con esta metodología, así como el Kernel(31); este algoritmo aporta a las aplicaciones de AA para el análisis de conjuntos de datos provenientes de información genética y genómica(44,45).
Los resultados obtenidos en este estudio demuestran que los algoritmos de AA tienen el potencial de generar predicciones útiles incluso bajo condiciones de información limitada, como el tamaño reducido de muestra y la baja densidad de marcadores genéticos. Este hallazgo resalta su aplicabilidad en escenarios prácticos donde los recursos son restringidos.
Sin embargo, se identificaron desafíos importantes, como el costo computacional y la dependencia de una cantidad suficiente de datos de calidad para maximizar la capacidad predictiva. A pesar de estas limitaciones, los algoritmos como RN y SVM mostraron un desempeño consistente, lo que sugiere que pueden ser herramientas valiosas en el análisis genómico. Estos resultados no solo brindan información práctica sobre el uso de los algoritmos de AA, sino que también abren la puerta a investigaciones futuras enfocadas en evaluar su comportamiento con bases de datos más amplias y detalladas, optimizando tanto su implementación como su capacidad predictiva en diferentes contextos.
Agradecimientos
Al Consejo Nacional de Humanidades, Ciencias y Tecnologías, México, por el financiamiento para el primer autor durante sus estudios de maestría. A la Asociación Mexicana de Criadores de Ganado Suizo de Registro por permitir el uso de su información.
Conflictos de interés
Los autores declaran que no existen conflictos de interés.
Literatura citada: