https://doi.org/10.22319/rmcp.v15i2.6366
Artículo
Estudio de la Estructura y Diversidad genética de ganado Holstein del sistema familiar en México
Felipe de Jesús Ruiz-López a
José G. Cortés-Hernández b
José Luis Romano-Muñoz a
Fernando Villaseñor-González c
Adriana García-Ruiz a*
a Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias (INIFAP). Centro Nacional de Investigación Disciplinaria en Fisiología y Mejoramiento Animal. Ajuchitlán Colón, 76280 Querétaro, México.
b Universidad Nacional Autónoma de México. Facultad de Medicina Veterinaria y Zootecnia. Ciudad Universitaria, Ciudad de México. México.
c INIFAP. Campo Experimental Centro-Altos de Jalisco. México.
*Autor de correspondencia: garcia.adriana@inifap.gob.mx
Resumen:
El objetivo fue conocer la estructura poblacional de los animales Holstein del sistema de lechería familiar, identificar posibles orígenes del material genético, conocer el grado de consanguinidad e identificar posibles huellas de selección en el genoma, que permitan vislumbrar las características que se han mejorado a través de los años. El estudio incluyó 270 animales genotipados con el chip GGP-50K®. Después del control de calidad de genotipos, se incluyeron 43,548 SNP autosómicos. Para conocer la estructura poblacional se realizaron análisis de mezclas y componentes principales (CP). Para conocer la consanguinidad genómica y detectar huellas de selección, se usó información de corridas de homocigosidad (ROH). El análisis de mezclas se realizó con el software Admixture, y los de CP, ROH y consanguinidad se realizaron con SVS-v7.6.8. El análisis de mezclas mostró evidencia de seis componentes, todos ligados a familias de sementales Holstein con diferente país de origen. Los CP no evidenciaron estratificación de la población por hato. El coeficiente de consanguinidad promedio fue de 0.59 ± 0.53 %. En las regiones del genoma con ROH más frecuentes en la población (≥20 animales), se han reportado numerosas asociaciones, QTL y genes relacionados con producción y composición de la leche, parámetros de fertilidad, susceptibilidad a enfermedades, conformación corporal, eficiencia alimenticia y algunas características de composición de la canal. Los resultados reflejan la existencia de una amplia diversidad genética en esta población y la posibilidad de realizar trabajos de mejoramiento genético a través de selección sin afectar los niveles de consanguinidad.
Palabras clave: Diversidad genética, Huellas de selección, Consanguinidad, Sistema de lechería familiar.
Recibido: 17/12/2022
Aceptado:09/05/2023
Introducción
La industria lechera de ganado bovino en México produjo alrededor de 11,489 millones de litros de leche a nivel nacional en el 2020 (SIAP, 2020)(1) de los cuales, más del 30 % del volumen se produjo en el sistema de lechería familiar (SLF), que incluye aproximadamente al 78 % de los establos(2). En los establos del SFL predominan los animales de la raza Holstein, aunque se pueden encontrar animales Pardo Suizo, así como las cruzas de estos(3). Actualmente en este sistema se cuenta con poca información de registros productivos por animal y en escasas ocasiones se puede recopilar información genealógica, lo que hace poco viable realizar evaluaciones genéticas de los animales de este sistema. El mejoramiento genético de estos animales se ha llevado a cabo por la selección que realiza el ganadero dentro de su hato, o por la introducción de material genético, pero no se tiene evidencia de apareamientos dirigidos con un fin genético determinado.
El uso de la información genómica ha permitido describir la estructura de las poblaciones que no cuentan con información genealógica o de registros. El estudio de estas poblaciones o animales se ha realizado a partir del estudio de marcadores de un solo polimorfismo (SNP) o los patrones de agrupamiento de los mismos; como, por ejemplo, las corridas de homocigosidad (ROH), que son segmentos homocigotos en el genoma, idénticos por descendencia, que pueden ser utilizados para estudiar la estructura de la población, la historia demográfica y para descifrar la estructura genética de enfermedades complejas(4). Las ROH son el resultado del cruzamiento entre individuos emparentados(5), provenientes de poblaciones con alto nivel de intensidad de selección, influenciada por la disponibilidad de reemplazos y adopción de herramientas tecnológicas y reproductivas(6), o bajas tasas de recombinación(7). Su distribución y longitud depende de la intensidad de la selección, siendo más frecuentes y más extensas cuando ésta es mayor(8), cuando los apareamientos entre parientes cercanos son frecuentes o cuando el tamaño de las poblaciones es reducida(4).
El potencial de las ROH para ayudar al mejoramiento genético de los animales de producción es grande, debido a que dentro de ellas se encuentran una gran cantidad de genes que codifican para características de interés(9). Además, la identificación de ROH puede ayudar a visualizar y reconocer patrones de haplotipos característicos de razas o especies(7), permitiendo identificar regiones genómicas con posibles huellas de selección para la raza(10) y calcular los niveles de consanguinidad individual. Esto último, mediante la evaluación de la porción del genoma cubierto por los segmentos ROH, especialmente porque que hay una alta probabilidad de detectar información genómica proveniente de parentescos antiguos(11). Herramienta útil para poblaciones que no cuentan con información genealógica(12).
Las huellas de selección son regiones del genoma que han sido conservadas por generaciones en las poblaciones debido a la selección natural o artificial. Estas secuencias de material genético están relacionadas con caracteres funcionalmente importantes(13) y su detección ayuda a identificar genes candidatos que se han favorecido en los procesos de selección a los que han sido expuestas las poblaciones, y a identificar mutaciones benéficas. Además, ayudan a la comprensión de las rutas moleculares relacionadas con los rasgos fenotípicos(14,15).
Con los análisis de marcadores tipo SNP, también es posible conocer la estructura poblacional a través de análisis de mezclas y conocer los orígenes más influyentes en una población. Además, mediante métodos reductivos de información, como lo son los análisis de componentes principales, es posible determinar patrones de la estructura poblacional, información importante para establecer las bases de un programa de mejoramiento genético.
El objetivo del presente estudio fue conocer la estructura poblacional, identificar posibles orígenes del material genético, conocer el grado consanguinidad e identificar posibles huellas de selección en el genoma, que permitan vislumbrar las características que se han mejorado a través de los años por las decisiones de los ganaderos en sistemas de producción familiar de México.
Material y métodos
Se utilizaron 270 genotipos de vacas Holstein, elegidas al azar de la población presente en tres hatos de SFL ubicados en la región de Tepatitlán, Jalisco, México. Los animales fueron genotipados con el chip GeneSeek Genomic Profiler Bovine GGP 50K®. El control de calidad a la información genómica consistió en excluir animales con tasa de llamada < 0.90; excluir SNP con frecuencia del alelo menor (MAF) < 0.02, o con una tasa de llamada < 0.95, o con un valor de P para Hardy Weinberg < 0.0001(16,17). Después del control de calidad, se incluyeron 43,548 SNP autosómicos.
Para conocer la estructura y principales orígenes poblacionales, se realizó un análisis de mezclas a través de la estimación basada en modelos de verosimilitud que definen la estructura de la ascendencia en individuos no relacionados, metodología implementada en el software Admixture V 1.3.0(18). Mientras que, para identificar posibles agrupaciones poblacionales por hato, se realizaron análisis de Componentes principales (CP). Para estimar la consanguinidad con información genómica y huellas de selección en la población, se buscaron ROH en el genoma. Para definir las ROH, se incluyeron las corridas que contaban con una longitud mínima de 500 kb y una cantidad mínima de 25 SNP, con una densidad mínima de 1 marcador cada 50 kb y una brecha máxima entre marcadores homocigotos contiguos de 500 Kb. Con los parámetros mencionados, se evitó el riesgo de incluir ROH muy cortas, caso común debido al desequilibrio de ligamiento (DL)(17). El DL se asocia con la presencia de genes ligados, por lo que al heredarse de padres a hijos lo hacen de forma conjunta, afectando la frecuencia de recombinación (menor del 50 %) y presencia de ROH en el genoma(19). Adicionalmente, se permitió 1 SNP heterocigoto y 5 genotipos perdidos por corrida(20,21).
El análisis de ROH se realizó con la plataforma bioinformática SNP & Variation Suite v7.6.8 Win64 (Golden Helix, Bozeman, MT, USA)(22), mientras que los análisis de los datos obtenidos se realizaron con SAS Institute 9.3.(23). Para analizar la distribución de LROH, se definieron seis clases, de acuerdo con su longitud, que fueron de 0.5 a 4, >4 a 8, >8 a 12, >12 a 16, >16 a 20 y >20 Mb(24).
Las huellas de selección se detectaron a través de la pérdida de variación genética, empleando las ROH identificadas en el genoma, utilizando las más frecuentes en la población (en al menos 10 % de los animales). De acuerdo con su posición física en el genoma, fueron identificadas anotaciones o regiones previamente relacionadas con genes, QTL o caracteres que se han realizado en otras poblaciones y que se encuentran reportadas en la base de datos Animal QTLdb Release 43(25). Además, se buscaron huellas de selección previamente reportadas en la base de datos de Variación del Genoma Bovino (BGVD)(26); lo anterior con la finalidad encontrar información genómica que ayude a conocer posibles características que se han seleccionado en la población del Sistema de lechería familiar (SLF) en México.
Para el cálculo del coeficiente de consanguinidad por corridas (FROH) se utilizó la metodología propuesta por Mcquillan et al(27), quienes la definieron como FROHi= ΣLROHi/ Lauto donde FROHi es el coeficiente de endogamia del individuo i calculado por ROH, ΣLROHi es la suma total de los segmentos de ROH de un individuo i por encima de una longitud mínima especificada, en este caso > 500 kb y Lauto es la longitud del genoma autosómico cubierta por los SNP incluyendo los centrómeros. Como el año de nacimiento de los animales es desconocido, se calculó la tendencia de consanguinidad por número de lactación de los animales al momento del muestreo.
Adicionalmente se realizó el cálculo del coeficiente de consanguinidad a través de marcadores homocigotos observados y esperados (FHOE) para todos los animales, el cual se ha reportado tiene una correlación de 0.96 ± 0.001 con la matriz de relaciones genómicas en ganado Holstein(28), los valores de FHOE pueden oscilar entre −1 y +1. Los números negativos se refieren a la exogamia presente en el apareamiento entre individuos de poblaciones diferentes y los valores positivos indican el nivel de endogamia de individuos de la misma población; el cálculo se realizó a través del método referenciado por Ferenčaković et al(11) con el programa SNP & Variation Suite v7.6.8 Win64 (Golden Helix, Bozeman, MT, USA)(22).
Resultados y discusión
En el análisis de mezclas, el valor que mejor definió el número de poblaciones ancestrales (K) fue seis y de acuerdo con la información recopilada de algunos padres de vacas, se lograron identificar seis familias grandes, definidas principalmente por el país de origen de los sementales. En la Figura 1-a, se muestra la estructura de la población ligada a los seis grupos principales por país de origen de los sementales, aunque algunas de esas familias comparten el mismo país de origen. Por lo anterior, se conjuntaron los grupos que compartieron el mismo origen, quedando sólo representados tres grupos grandes, dos que representan a Estados Unidos de América y uno al Reino Unido (USA, 840 y GBR, respectivamente) (Figura 1-b). Los orígenes USA y 840 corresponden a Estados Unidos de América, sólo que el 840 es asignado a los animales que usan identificaciones de radiofrecuencia (RFID), dispositivos expedidos por el Comité Internacional de Registro Animal (ICAR); mientras que los animales registrados con país de origen USA no llevan un RFID y el uso del material genético es a nivel local o más limitado que los 840(29). Los resultados de este estudio evidencian la dependencia genética que tiene el sistema familiar en México del material extranjero, principalmente proveniente de Estados Unidos, ya que más del 80 % de los orígenes fueron ligados a familias con orígenes de este país, ya sea de comercio local o de los registrados internacionalmente.
Figura 1: Estructura poblacional del ganado Holstein del sistema familiar a) incluyendo los seis orígenes atribuidos a familias de sementales y b) agrupados por país de origen
A pesar de que en otro estudio(29) se había reportado la influencia de otras razas de bovinos lecheros en el sistema familiar, en el presente estudio no se encontró evidencia del uso o cruza con otras razas. Estos resultados podrían sugerir que los ganaderos han seguido un sistema de apareamientos más dirigidos, y que se limitan a usar animales de la misma raza en los servicios.
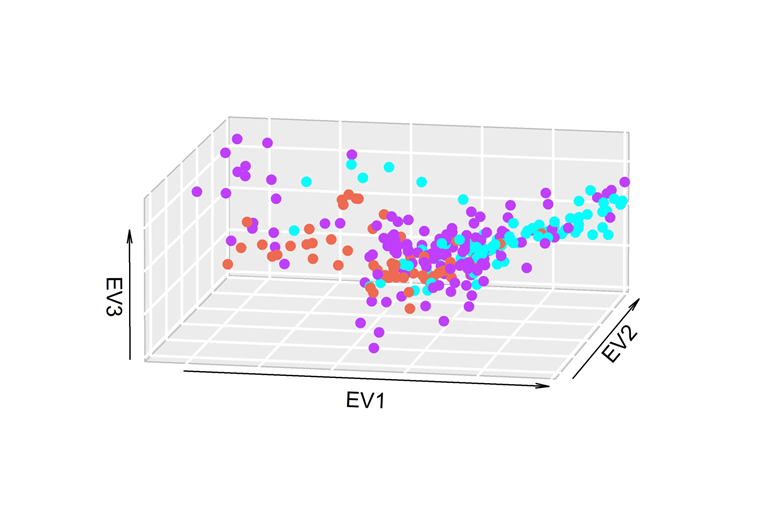
En los análisis de CP (Figura 2), no se encontró estratificación por país de origen del semental, y cuando se evaluó el hato de origen, se observa un hato (morado) homogéneo en la población, y se aprecia una diferencia entre los animales de los otros hatos (rojo y azul). El porcentaje de la variabilidad asociada a cada componente fue de 2.9, 2.0 y 1.8 para los componentes o eigenvalores (EV) 1, 2 y 3 respectivamente.
Figura 2: Análisis de componentes principales de la población del sistema familiar con fenotipo Holstein, definidos por el hato de origen
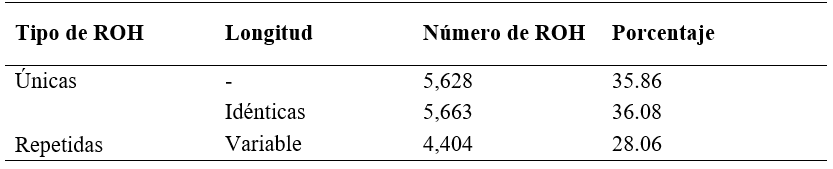
El número total de ROH (NROH) encontrado en la población estudiada fue 15,695 con una longitud promedio (LROH) de 4.79 Mb, una longitud mínima y máxima de 0.5 y de 91.49 Mb, respectivamente. La longitud promedio del genoma cubierto por ROH fue de 278.76 Mb, con un mínimo y máximo de 13.28 y 535.83 Mb, respectivamente. De acuerdo con la frecuencia de las ROH en la población, se identificaron como únicas (en un solo animal) o repetidas, estas últimas con una misma longitud (idénticas) o de longitud variable. El 35.86 % de las ROH fueron únicas (Cuadro 1); mientras que el 64.14 % (10,067) fueron repetidas.
Cuadro 1: Número y porcentaje de corridas de homocigosidad (ROH) únicas y ROH repetidas con misma posición de inicio y fin, así como posiciones variables
El NROH fue menor en comparación con lo reportado en animales que provienen de sistemas especializados de producción, lo que podría atribuirse a una menor intensidad de selección, ya que la pérdida de variación genética o la formación de ROH en el genoma se encuentra influenciada, entre otros factores, por el nivel de intensidad de selección en las poblaciones, que a su vez está determinado por la disponibilidad de reemplazos y adopción de herramientas tecnológicas y reproductivas, como lo son la inseminación artificial (IA) y la transferencia de embriones (ET). En ganado lechero, la intensidad de selección es muy alta y la selección de material genético se encuentra influenciada por un número limitado de familias progenitoras, por lo que el apareamiento de individuos emparentados puede ser común(30). En el ganado Holstein del sistema especializado de producción en México, el NROH fue de 88,529, con un tamaño de la población mayor (~4,500 animales) y LROH fue mayor a 8.95 Mb(24). En otros estudios en ganado Holstein del sistema especializado las LROH reportadas son aún mayores; por ejemplo, en EUA de 299.6 Mb(31) y en Italia de 297 Mb(32).
El número promedio de ROH por animal fue de 58.13 ± 11.89, con un máximo y mínimo de 92 y 10; lo que es un valor alto comparado con los resultados de ganado Holstein del sistema especializado en México(24) reportado en promedio en 20.07 ROH por animal con un máximo de 283 y un mínimo de 1. Estudios en otras poblaciones Holstein de producción intensiva han reportado alrededor de 82.3 ± 9.83 ROH por animal en ganado Holstein de EUA(31) y 81.7 ± 9.7 corridas por animal en ganado Holstein de Italia(32). Estas diferencias pueden ser debidas al alto grado de selección en las poblaciones de sistemas de producción especializados tanto de EUA como de Italia, así como a la disponibilidad de material genético de sementales altamente seleccionados en comparación con el SLF que se analizó en donde no están tan definidos los objetivos de selección.
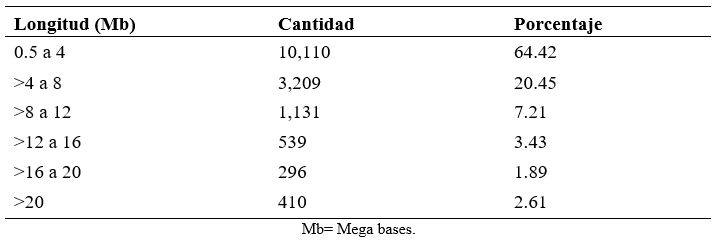
De acuerdo con la clasificación de LROH, las corridas más frecuentes fueron las más cortas (0.5 a 4 Mb) con el 64.42 %, seguidas de las de 4 a 8 Mb con 20.45 %, y las menos frecuentes fueron las corridas más largas (>16 a 20 y >20; Cuadro 2). Las longitudes de las ROH proporcionan información sobre el número de generaciones en las que se comparte el ancestro común, siendo las más largas las formadas en generaciones recientes(21), por lo que la longitud de las ROH encontradas en esta población reflejan una consanguinidad reciente y baja.
Cuadro 2: Frecuencia de corridas de homocigosidad (ROH) en distintas longitudes
El coeficiente de consanguinidad (FROH) promedio en la población fue de 0.59 ± 0.53 % con un máximo de 3.35 % y un mínimo de 0.034 %. Los resultados concuerdan con la poca cantidad de ROH encontradas y la corta longitud promedio. Estos valores se encontraron muy por debajo a lo reportado en otras poblaciones de ganado Holstein altamente seleccionadas; por ejemplo, 4.2 % en EUA(33). Aunque la consanguinidad encontrada en esta población fue insignificante, valor que se confirma con los valores calculados para FHOE que fueron de -0.02 ± 0.08, al revisar los promedios por número de parto, se encontró un leve incremento en FROH de las generaciones recientes, lo que podría indicar el inicio de una tendencia desfavorable para el grupo estudiado (Figura 3).
Figura 3: Porcentajes de consanguinidad genómica (FROH) por número de lactación al momento del muestreo
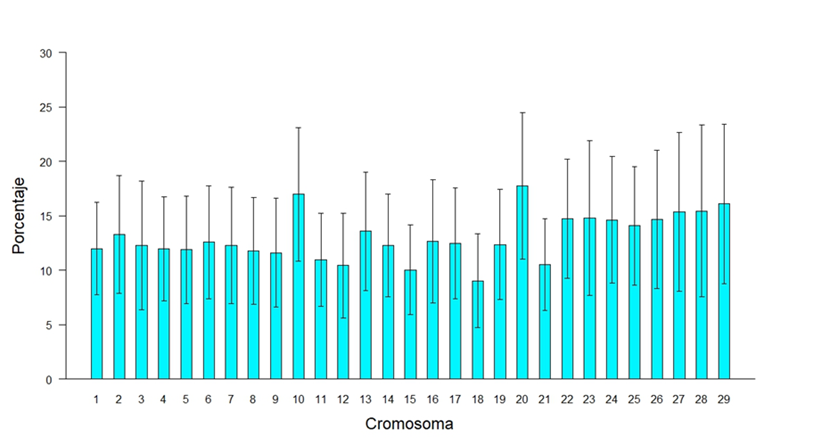
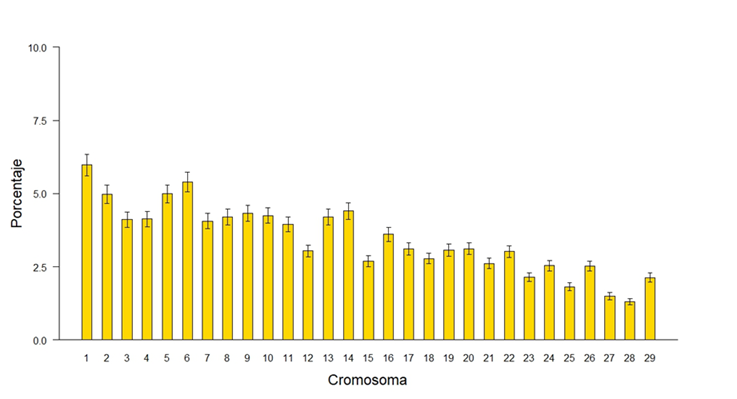
Para identificar las huellas de selección a lo largo del genoma, se buscaron cromosomas y regiones específicas en la ubicación y distribución de las ROH. La presencia de ROH se mostró en mayor medida en los cromosomas largos que en los cortos, aunque estos últimos presentaron una mayor proporción del genoma cubierta por regiones homocigóticas como lo fue en el caso de los cromosomas10 y 20, que presentaron un 10 y un 20 con 16.98 % y 17.76 % (Figura 4), comportamiento similar a lo reportado por Szmatola et al(34) en vacas Holstein de Polonia sugiriendo que estas regiones han estado sujetas a una mayor selección, por asociación a caracteres de interés económico. Los porcentajes de homocigosidad por cromosoma son mayores que el valor promedio de FROH porque se toma la longitud del cromosoma como el ciento por ciento y no la longitud total del genoma cubierto por los SNP; con esto se tiene una mejor percepción de la longitud del cromosoma cubierto por ROH.
Figura 4: Porcentaje del cromosoma cubierto por corridas de homocigosidad (ROH)
A lo largo del genoma se observó una relación positiva entre el tamaño de cromosoma y el número de ROH detectadas en ese cromosoma, pero no fue así para el porcentaje de la longitud del cromosoma cubierto por ROH, ya que los cromosomas cortos mostraron una mayor proporción cubierta por ROH (Figura 5), esto debido a que la longitud promedio de las ROH fue mayor en los cromosomas cortos que en los largos, debido a que en los cromosomas largos existe mayor recombinación que en los cromosomas cortos(8). Del total de ROH determinado en la población, los cromosomas 1 y 6 fueron los que presentaron una mayor cantidad de ROH (5.98 y 5.39 %) y los cromosomas con una menor cantidad fueron los cromosomas 28 y 27 (1.31 y 1.50 %) resultados similares a los de Purfield et al(17)quienes también reportaron una mayor cantidad de ROH en los cromosomas largos que en los cortos.
Figura 5: Porcentaje de corridas de homocigosidad (ROH) en cada cromosoma
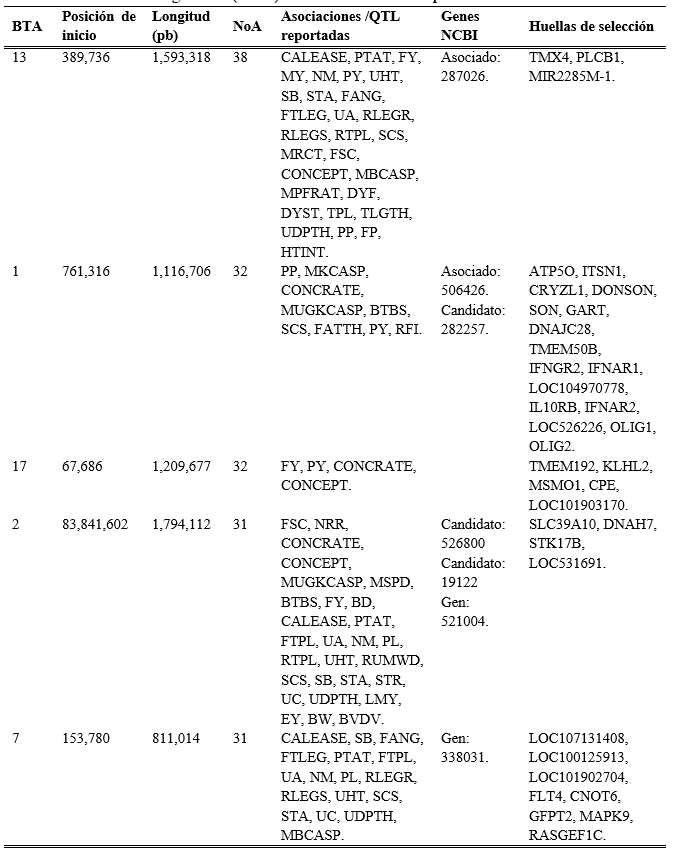
De acuerdo con la frecuencia de las ROH repetidas en la población, sólo 35 se encontraron en 10 animales o más y se distribuyeron en los cromosomas 2, 3, 4, 5, 6, 8, 9, 10, 11, 12, 13, 14, 15, 17, 19, 21, 22, 23, 26 y 29. Las ROH más frecuentes se encontraron en cromosomas 2 y 22, en 27 y 23 animales, respectivamente, siendo la longitud en estas corridas de 1.82 Mb y 1.61 Mb. En la misma posición de las corridas encontradas en el cromosoma 2 (83.84-85.66 Mb), Cole et al(35) dieron a conocer QTL (Loci de Rasgos Cuantitativos) relacionados con el ancho de la cadera y estatura en ganado Holstein de EUA; Cai et al(36) reportaron QTL asociados a producción de grasa en leche en ganado Holstein de países Nórdicos, pudiendo indicar huellas de selección en estos cromosomas(37).
Del total de ROH con longitud variable, sólo 37 se encontraron en 10 animales o más, distribuyéndose a lo largo del genoma, excepto en el cromosoma 8. En la región donde se encontraron las ROH más frecuentes en la población (≥20 animales), se han reportado numerosas asociaciones, QTL y genes que se encuentran relacionados con producción y composición de la leche, parámetros de fertilidad, susceptibilidad a enfermedades, conformación corporal, eficiencia alimenticia y algunas características de composición de la canal (Cuadro 3). Los resultados muestran que a pesar de que las longitudes de las ROH en esta población (~4.79 Mb) sugieren un cruzamiento de animales emparentados hace aproximadamente 16 generaciones(11), las regiones conservadas podrían indicar que la selección en esta población se encuentra dirigida a mejorar la producción de leche, composición, fertilidad y salud, como podría esperarse en los sistemas de producción de leche. En los cromosomas 1 y 2, además de las asociaciones con características de interés en ganado lechero, se observan asociaciones con características de la canal, hallazgos que pudieran sugerir posibles cruzamientos con otras razas.
Para buscar anotaciones de ROH con longitud variable, se tomó como referencia la ROH más corta con respecto a la posición final (Cuadro 3), para evitar proporcionar información fuera de la región común a todos los animales con una ROH específica.
Cuadro 3: Anotaciones en el genoma encontradas en las regiones donde se detectaron las corridas de homocigosidad (ROH) más frecuentes en la población de lechería familiar
BTA= cromosoma, NoA= número de animales.
Asociaciones /QTLs reportados. CALEASE= facilidad de parto, PTAT= puntos finales de conformación, FY= producción de grasa en leche, MY= producción de leche, NM= mérito Neto, PY= producción de proteína en leche, UHT= altura de inserción de la ubre, SB= mortinatos, STA= estatura, FANG= ángulo de la pezuña, FTLEG= conformación de patas y pezuñas, UA= inserción de la ubre, RLEGR= colocación de las patas traseras - vista trasera, RLEGS= vista lateral de aplomos posteriores, RTPL= posición de tetas posteriores, SCS= puntuación de células somáticas, MRCT= tiempo de coagulación del cuajo de la leche, FSC= concepción a primer servicio, CONCEPT= número de inseminaciones por concepción, MBCASP= porcentaje de B-Caseína en leche, MPFRAT= proporción de proteína y grasa de leche, DYF= carácter lechero, DYST= distocia, TPL= posición de tetas, TLGTH= longitud de pezones, UDPTH= profundidad de la ubre, PP= porcentaje de proteína en leche, FP= porcentaje de grasa en leche, HTINT= intensidad del estro, MKCASP= porcentaje de Kappa caseína en leche, CONCRATE= tasa de concepción, MUGKCASP= porcentaje de kappa caseína en leche no glucosilada, BTBS= susceptibilidad a tuberculosis bovina, FATTH= espesor de grasa en la 12a costilla, RFI= consumo residual, NRR= tasa de no retorno, MSPD= velocidad de ordeño, BD= profundidad de Cuerpo, FTPL= posición de tetas anteriores, PL= duración de vida productiva, RUMWD= anchura del anca, STR= fortaleza lechera, UC= hendidura de ubre, LMY= rendimiento de carne magra, EY= energía de producción láctea, BW= peso corporal al nacimiento, BVDV= susceptibilidad a diarrea viral bovina.
Genes NCBI. 287026= fosfolipasa C beta 1, 506426= crystallin zeta codificador de proteínas, 282257= subunidad 1 del receptor de interferón alfa y beta, 526800= ankyrin repetidor dominio 44, 19122= proteína priónica, 521004= portador de soluto familia 39 miembro 10, 338031= receptor relacionado con fms tirosina quinasa 4.
Huellas de selección (genes codificadores de proteínas). TMX4= proteína transmembrana 4 relacionada con tiorredoxina, PLCB1= fosfolipasa C beta 1, MIR2285M-1= microARN que participan en la regulación postranscripcional de la expresión génica, ATP5O= subunidad del tallo periférico de la ATP sintasa, OSCP= subunidad periférica del tallo de ATP sintasa, ITSN1= intersección 1, CRYZL1= crystallin zeta codificador de proteínas, DONSON= factor de estabilización de la horquilla de replicación del ADN, SON= proteína de unión a ADN y ARN, GART= fosforribosilglicinamida formiltransferasa y sintetasa, fosforribosilaminoimidazol sintetasa, DNAJC28= familia de proteínas de choque térmico, TMEM50B= proteína transmembrana 50B, IFNGR2= receptor de interferón gamma 2, IFNAR1= subunidad 1 del receptor de interferón alfa y beta, LOC104970778= gen ARN no caracterizado, IL10RB= subunidad beta del receptor de interleucina, IFNAR2= subunidad 2 del receptor de interferón alfa y beta, LOC526226= histona H4, OLIG1= factor de transcripción de oligodendrocitos 1, OLIG2= factor de transcripción de oligodendrocitos 2, TMEM192= proteína transmembrana 192, KLHL2= kelch de la familia 2, MSMO1= metilsterol monooxigenasa 1, CPE= carboxipeptidasa E, LOC101903170= cen ARN no caracterizado, SLC39A10= familia de portadores de soluto 39, DNAH7= cadena pesada axonemal de dineína 7, STK17B= serina/treonina quinasa 17b, LOC531691= dominio HECT, C2 y WW que contiene la proteína ligasa 2 de ubiquitina E3, LOC107131408= familia de receptores olfativos 5 subfamilia W miembro 39, LOC100125913= gen no caracterizado, LOC101902704= familia de dominios de lectina de tipo C, 7, A, FLT4= receptor relacionado con fms tirosina quinasa 4, CNOT6= subunidad 6 del complejo de transcripción CCR4-NOT, GFPT2= glutamina-fructosa-6-fosfato transaminasa 2, MAPK9= proteína quinasa 9 activada por mitógeno, RASGEF1C= miembro de la familia de dominio RasGEF 1C.
En la población de estudio, se identificaron ROH que se han mantenido como resultado del proceso de selección de la población del sistema familiar. Estas regiones conservadas, se encuentran asociaciones de marcadores tipo SNP, QTL y genes; que en su mayoría están relacionados con características de interés económico en la industria lechera, como producción y composición de la leche, parámetros de fertilidad, susceptibilidad a enfermedades, conformación corporal, eficiencia alimenticia y algunas otras características como la composición de la canal, lo que podría tomarse como huellas de selección (Cuadro 3). Estos resultados muestran los caracteres que han sido incluidos en los procesos de selección en la población; ya sea de forma intencional por la selección que realizan los ganaderos o no intencional por la disponibilidad de material genético en el mercado, ya que, al usar la IA, la elección de sementales guía al ganadero a modificar la genética de sus animales en la forma que lo hacen las empresas de IA.
Conclusiones e implicaciones
El ganado productor de leche del SLF tiene orígenes ancestrales de países que son proveedores de material genético a nivel internacional, como son EE.UU. y GBR, sin mostrar evidencia de cruzamientos recientes con otras razas lecheras. Dentro de la población estudiada, se puede observar genéticamente homogénea, con una menor cantidad y longitud de ROH que los animales en sistemas especializados de producción, lo que refleja una amplia variación genética causada por una baja intensidad de selección. En este trabajo se identificaron ROH que se han mantenido como resultado del proceso de selección, que en su mayoría están relacionados con características de interés económico en la industria lechera.
Los resultados de este estudio reflejan la existencia de un bajo nivel de consanguinidad en la población y una mayor diversidad genética en esta población respecto a los que se encuentran en sistemas especializados, por lo que se tiene la posibilidad de realizar trabajos de mejoramiento genético dirigidos a las características de interés de los productores a través de selección, sin que la consanguinidad comprometa la productividad y salud de la población.
Agradecimientos y fuente financiadora
Proyecto financiado por INIFAP-CENIDFyMA con el nombre “Desarrollo de una estrategia integral sustentable para incrementar la disponibilidad de reemplazos Holstein de buena calidad en el sistema familiar de producción de leche en México” con No SIGI: 15352034772.
Conflicto de intereses
Los autores declaran que no existen conflictos de interés.
Literatura citada: