https://doi.org/10.22319/rmcp.v13i4.6138
Article
Exploring bovine fecal bacterial microbiota in the Mapimi Biosphere Reserve, Northern Mexico
Irene Pacheco-Torres a
Cristina García-De la Peña b*
César Alberto Meza-Herrera a
Felipe Vaca-Paniagua c,d,e
Clara Estela Díaz-Velásquez c
Claudia Fabiola Méndez-Catalá c
Luis Antonio Tarango-Arámbula f
Luis Manuel Valenzuela-Núñez b
Jesús Vásquez-Arroyo g
a Universidad Autónoma Chapingo. Unidad Regional Universitaria de Zonas Áridas, Bermejillo, Durango, México.
b Universidad Juárez del Estado de Durango. Facultad de Ciencias Biológicas, Av. Universidad s/n Fracc. Filadelfia, 35010 Gómez Palacio, Durango, México.
c Facultad de Estudios Superiores Iztacala. Laboratorio Nacional en Salud, Diagnóstico Molecular y Efecto Ambiental en Enfermedades Crónico-Degenerativas. Tlalnepantla, Estado de México.
d Instituto Nacional de Cancerología. Ciudad de México, México.
e Universidad Nacional Autónoma de México. Facultad de Estudios Superiores Iztacala Unidad de Biomedicina, Tlalnepantla, Estado de México, México.
f Colegio de Postgraduados, Campus San Luis Potosí, Salinas de Hidalgo, San Luis Potosí, México.
g Universidad Juárez del Estado de Durango. Facultad de Ciencias Químicas. Gómez Palacio, Durango, México.
* Corresponding author: cristina.garcia@ujed.mx
Abstract:
In Mexico, information on the bovine fecal microbiota (Bos taurus) is scarce. The present study describes the diversity and abundance of bacteria in fecal samples from rangeland bovines, collected in the Mapimi Biosphere Reserve in the central part of the Chihuahuan desert. Fecal samples were analysed using high-throughput next generation massive sequencing using V3-V4 16S rRNA on Illumina Miseq. A total of 17 phyla, 24 classes, 33 orders, 50 families, 281 genera, and 297 species were identified. Firmicutes and Verrucomicrobia were the most abundant phyla. The most abundant genera were Sporobacter, PAC000748_g (genera into the Ruminococcaceae family) and Eubacterium_g23. Three genera (Clostridium, Corynebacterium and Fusobacterium) and one species (Campylobacter fetus) potentially pathogenic bovine bacteria were registered. This information represents a bacteriological baseline for monitoring the grazing bovine intestinal health status, and to trace possible interactions with the fecal microbiota of native roaming wildlife in the area.
Key words: Bos taurus; Campylobacter fetus; Bacterial diversity; 16S rRNA gene; Massive sequencing.
Received: 13/01/2022
Accepted: 06/04/2022
Introduction
The microbial community of the gastrointestinal system of cattle remains understudied. Due to its influence on nutrient absorption, productivity, potential reservoir of human and animal pathogens, as well as overall animal health, there is a need to better understand bovine gut microbial communities(1). Recently, high-throughput sequencing using 16S rRNA amplicons has provided deeper information on the fecal bovine microbiota composition, and the results obtained to date indicate a high diversity(2).
The central part of the Chihuahuan Desert in Mexico has a high diversity of wildlife(3,4). The bovine (Bos taurus) has been raised as grazing livestock since its introduction at the end of the 16th century, being the most important economic activity in this area(5). However, this activity is the main reason of ecological deterioration which affects wildlife; for example, this ruminant species competes for forage resources with endemic animal species (i.e., Gopherus flavomarginatus, Bolson tortoise)(3). Cattle grazing also exerts strong pressure on plant populations, modifying their cover; this may increase soil erosion susceptibility in this desert(3,6).
The cattle gut microbiome has many microbial species that play an important role in health and productivity(7,8). These microbes are essential for the fermentation of consumed plant matter that is converted into energy for the host(9). However, bovines asymptomatically transport bacterial species that are potential pathogens to wildlife as Escherichia coli, Campylobacter spp., Salmonella spp. and Listeria spp.(6,10). In recent years, the extensive use of land for agriculture has increased the densities of cattle populations creating positive correlations with pathogenic infections by fecal bacteria(11). Though, knowledge about bovine fecal bacterial diversity under grazing management systems is relatively scarce(12). This study aimed to explore for the first time the diversity and abundance of fecal bacteria from bovines under grazing-marginal conditions in the Mapimi Biosphere Reserve, center of the Chihuahuan desert, using next-generation sequencing (16S rRNA).
Material and methods
All the methods and activities of this study were in strict accordance with accepted guidelines for ethical use, care and welfare of animals in research at international(13) and national(14) levels, with institutional approval reference number UJED-FCB-2018-07.
Study area
The study was developed in the Mohovano de las Lilas locality, northeast of the Mapimi Biosphere Reserve in Mexico (26°00’ and 26°10’N, 104°10’ and 103°20’W) in the center of the Chihuahuan desert. This area has warm, very arid climate, with an average annual temperature of 25.5 ° C, and an average annual precipitation of 264 mm. The predominant vegetation is rosette and microphile scrub, as well as halophyte, and gypsophila plants(15).
Field work
In July 2018, three fresh fecal samples were collected from three healthy male bovines. From each fecal sample, 0.25 g was collected from the center of the sample and deposited it in BashingBead™ cell lysis tubes (Zymo Research Corp.) adding 750 μL of lysing/stabilizing solution. Each tube was processed in a TerraLyzer™ cellular disruptor (Zymo Research Corp.) during 20 sec according to the equipment specifications.
Laboratory work
DNA was extracted from the samples using the Xpedition™ Soil/Fecal DNA MiniPrep kit (Zymo Research Corp.) in a laminar UV flow hood in sterile conditions. The amount of DNA obtained was measured in a Qubit™ fluorometer (Invitrogen). Then, the V3-V4 region of the 16S rRNA gene was amplified using the following primers(16): S-D-Bact-0341-b-S-17, 5´-CCTACGGGNGGCWGCAG-3´ and S-D-Bact-0785-a-A-21, 5´-GACTACHVGGGTATCTAATCC-3´. The step after sequencing was realized using a Illumina protocol(17,18) and thereafter, the samples was sequenced in MiSeq of 2 × 250 paired final. The complete sequencing process is available in García-De la Peña et al(19).
Data availability
The files used in this study were deposited into the NCBI Sequence Read Archive (SRA) database (Accession Number: PRJNA614584).
Bioinformatic analysis
The DNA sequences were analyzed using Quantitative Insights into Microbial Ecology bioinformatics software (QIIME)(20). Both forward and reverse sequences were assembled using the PEAR program(21) considering Q30 the quality criterion (one false base for every 1,000 bases). Chimeric sequences were discarded with USEARCH(22). Then, the operational taxonomic units (OTUs) were selected with the UCLUST method(22) at 97 % similarity; a representative sequence for each OTU was obtained, and the taxonomy was assigned using EzBioCloud database as reference(23). A simple random rarefaction process was performed(24) in order to obtain a standardized file for all samples. The relative abundance for the phylum and family levels were represented as stacked bar plots using R, and genus level was visualized as a heatmap using Morpheus software (Morpheus, https://software.broadinstitute.org/morpheus); hierarchical clustering (average linkage method with Euclidean distance) was used to visualize samples dendrogram(25).
Results and discussion
In this study, the average number of sequences assembled was 155,915. A mean ± sd of 109,814 ± 16,686 bacterial sequences were obtained after taxonomic designation. The average number of OTUs with a 97 % of similarity was 6,661 ± 431 (Table 1).
Table 1: Fecal sequences information of Bos taurus at the Mohovano de las Lilas locality, Mapimi Biosphere Reserve, Mexico
Sample | Total | Assembled | Discarded | BS | BSS | OTUs |
1 | 322,428 | 138,862 | 183,566 | 131,275 | 98,084 | 6,293 |
2 | 223,470 | 145,379 | 78,091 | 136,807 | 102,441 | 6,556 |
3 | 305,380 | 183,506 | 121,874 | 173,177 | 128,916 | 7,135 |
Mean | 283,759 | 155,915 | 127,843 | 147 086 | 109 814 | 6,661 |
BS= bacteria sequences after taxonomical designation, BSS= bacteria sequences after singletons removal; OTUs= operational taxonomic units.
A total of 17 phyla, 24 classes, 33 orders, 50 families, 281 genera, and 297 species were determined. The most abundant phyla (Figure 1) were Firmicutes (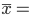 88.9 %) and Verrucomicrobia ( 6.4 %). The same phyla were reported in grazing Mongolian cattle in Hulunbuir grassland and Alxa Desert in China(26). These phyla are considered normal components in the basic fecal microbiota of domestic herbivores(27,28) and other species of ruminants(29,30). Firmicutes has been reported as the most frequent phylum in fecal samples of cattle, horses(2,31,32), and red deer(33). This abundance is related to high fiber intake(34). Verrucomicrobia was the second abundant phylum in the cattle samples in this study. Aricha et al(26) determined that this phylum was very abundant in the intestinal tract of the grazing Mongolian cattle in the Alxa Desert, and argue that this may be related to the extremely strong disease resistance of this breed of cattle. It is important to analyze later if this phylum confers resistance to cattle diseases in the Mapimi reserve, which would represent an advantage for the bovine’s health in this area. Also, Bacteroidetes was reported in previous studies Mongolian(26), and Holstein Friesian(36) as the second most abundant phylum in other cattle species such as grazing and feedlot Angus Beef(35). However, Bacteroidetes was found in a minimum proportion (0.001%) in the cattle samples of the Mapimi reserve. This disparity can be related to the type of diet(37), geographical differences(26), and the environment in which they are distributed(38). Nevertheless, this information can only be confirmed by developing specific studies in this respect.
88.9 %) and Verrucomicrobia ( 6.4 %). The same phyla were reported in grazing Mongolian cattle in Hulunbuir grassland and Alxa Desert in China(26). These phyla are considered normal components in the basic fecal microbiota of domestic herbivores(27,28) and other species of ruminants(29,30). Firmicutes has been reported as the most frequent phylum in fecal samples of cattle, horses(2,31,32), and red deer(33). This abundance is related to high fiber intake(34). Verrucomicrobia was the second abundant phylum in the cattle samples in this study. Aricha et al(26) determined that this phylum was very abundant in the intestinal tract of the grazing Mongolian cattle in the Alxa Desert, and argue that this may be related to the extremely strong disease resistance of this breed of cattle. It is important to analyze later if this phylum confers resistance to cattle diseases in the Mapimi reserve, which would represent an advantage for the bovine’s health in this area. Also, Bacteroidetes was reported in previous studies Mongolian(26), and Holstein Friesian(36) as the second most abundant phylum in other cattle species such as grazing and feedlot Angus Beef(35). However, Bacteroidetes was found in a minimum proportion (0.001%) in the cattle samples of the Mapimi reserve. This disparity can be related to the type of diet(37), geographical differences(26), and the environment in which they are distributed(38). Nevertheless, this information can only be confirmed by developing specific studies in this respect.
Figure 1: Relative abundance (%) of fecal bacteria taxa (phylum level) from three samples of Bos taurus at the Mohovano de las Lilas locality. Only the first 10 more abundant phyla are shown
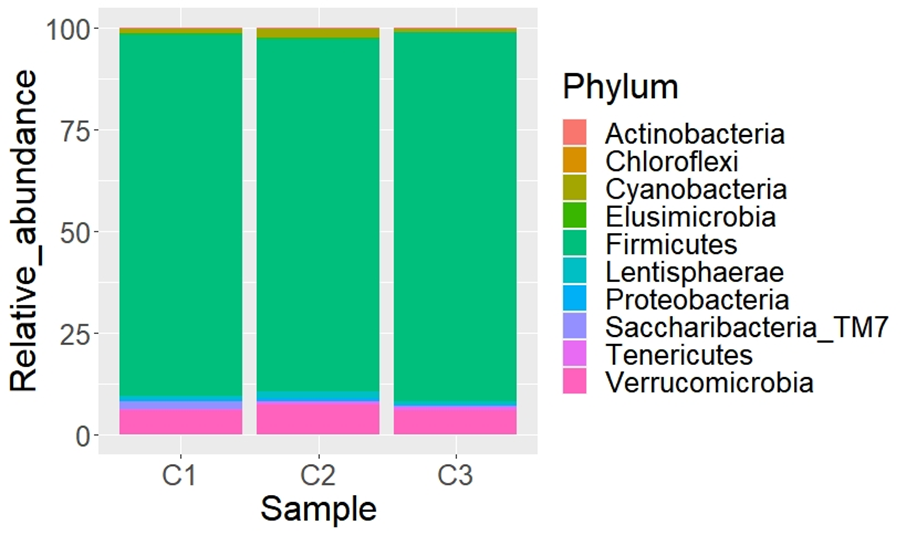
At family level, Ruminococcaceae ( 68.9 %) and Lachnospiraceae ( 10.9 %) were abundant in the fecal samples collected (Figure 2); both families are found in the mammalian gut environment and have been associated with good health(39). Some genera of the Ruminococcaceae family are part of the normal intestinal microbiota of cattle, sheep, and goats metabolizing cellulose, and colonizing the rumen(40); these bacteria taxa are important for the degradation and fermentation of polysaccharides in the diet of ruminants(41). In addition, it has been reported that members of the Lachnospiraceae family exhibit pectin hydrolysis activities in the cattle´s rumen(42) associated to the butyric acid production and providing energy for the growth of intestinal epithelial cells(43). The high abundance of Lachnospiraceae in cattle protects the intestine and acts as a barrier that favors the adaptation of the host to its environment; it also promotes a decrease in the incidence of intestinal diseases(26).
Figure 2: Relative abundance (%) of fecal bacteria taxa (family level) from three samples of Bos taurus at the Mohovano de las Lilas locality. Only the first 10 more abundant families are shown
From 281 classified genera found in this study, 36.6 % have a taxonomic name; this percentage is higher than the reported by Kim and Wells(44) in feces of cattle where only 110 genera were classified, and about 41 % of the total sequences couldn’t be assigned to a known genus (Figure 3). Nevertheless, the results showed here increase the number of genera of the B. taurus fecal microbiota previously reported (12,45,46), who confirmed that the fecal bacterial microbiota is extremely diverse in cattle, and has not yet fully described. Sporobacter was the most abundant genus found in the fecal samples of cattle in this study. This genus was reported in alpaca(47), deer sika(28), horse(48), donkey(49), and the Bolson tortoises Gopherus flavomarginatus(19). This genus is related to digestion of plant ligno-cellulosic matter; however relatively little is known about the role of this bacteria in the degradation process(50). Durso et al(51) reported Faecalibacterium, Ruminococcus, Roseburia, and Clostridium as important components of the fecal bovine microbiota. These genera were also determined in the present study. According to some studies(52,53) these bacteria constitute 50 to 70 % of the total number of microorganisms in the digestive system of ruminants. These animals have specific gut microbial taxa as they are dependent on these bacteria to extract energy and nutrients from food(54), besides having specialized anatomical and physiological adaptations to the cellulolytic fermentation of low nutrition - high fiber vegetal material(55). The presence of other bacterial genera reported in this study could be the result of environmental and genetic factors, age, breed, diet, phylogeny, among others(56,57,58). Recently(56,59,60), was demonstrated that herbivorous animals have the most diverse microbiota since they depend on microbial metabolic pathways to maximize energy and nutrient extraction from feeding(61).
Figure 3: Heatmap of Bos taurus fecal bacteria sample at genus level at the Mohovano de las Lilas locality. Only the first 40 more abundant genera are shown

Although the gut microbiome usually remains stable over time assisting as a defense system against pathogens and other disease-causing agents in the host, the disturbance of this community can lead to animal disease(62,63). In the present study, the samples collected were obtained from apparently healthy bovines. However, bacteria considered of veterinary importance were found in these animals; this could be a potential health risk because they are carriers of these microorganisms. For example, Campylobacter, Clostridium, Corynebacterium and Fusobacterium were found in the fecal samples. These genera have been associated with cattle disease. Campylobacter has been reported as a cause of infertility and abortion in ruminants(64,65); also represents a critical threat to public health, because it can be transmitted from cattle to humans(66,67,68). Clostridium has been reported causing diseases and death in ruminants, especially in cattle; examples are respiratory diseases(69), botulism(70) and the blackleg(71). Corynebacterium has been reported in beef and dairy cattle associated with renal disease(72), mastitis(73,74), and tuberculosis(75,76,77); also, it is considered as an important emergent pathogen for humans(78). Finally, Fusobacterium was reported by others(79-82), causing abscesses in cattle. It is important to develop other studies that provide information on the pathogenicity and dynamics of these potential pathogens in the bovines of the Mapimi reserve.
At species level, Pseudobacteroides cellulosolvens and Campylobacter fetus were registered in the present study. Pseudobacteroides cellulosolvens is anaerobic bacteria that degrade plant cell wall polysaccharides and cellulosic, being capable of using cellulose or cellobiose as a sole carbon source(83). Campylobacter fetus is a relevant species; the main reservoirs of this bacteria are both the intestinal and the genital tracts in cattle and sheep(64,65). This species causes spontaneous abortion and infertility in cattle, while it is also an opportunistic pathogen to humans(84).
Due to the free-grazing management in the Mapimi Reserve, the bovine feces remain over the soil until natural processes degrade them. Consequently, native fauna can be in contact with these feces, increasing the probability of interspecific transmission of some bacteria(85). Although it has been previously reported that there are no evidence of cross-parasite infection between cattle and mule deer in the Mapimi Biosphere Reserve(86), it is important to clarify whether this same scenario occurs for bacteria. McAllister and Topp(87) estimate that about 77 % of the pathogens that usually infect livestock can also affect wildlife. However, also wildlife is considered an important source of microorganisms that could cause infectious diseases to domestic animals and humans(88,89). For these reasons, it is important to develop studies focused on risk management at the interface of domestic species and native fauna, considering the implications for the transmission of microorganisms with pathogenic potential(88,89). This information could lead to establish microbiological control strategies for wild fauna populations and livestock within the area.
Conclusions and implications
Information about the bovine fecal microbiota under extensive grazing conditions is scarce. From economic, ecological and health perspectives, it is crucial to determine the bacterial diversity -from phyla to species-, in the intestine of domestic ruminants. The present study is the first insight into the fecal bacterial composition of bovines in the Mapimi Biosphere Reserve in Mexico using next generation sequencing. This information significantly expands the knowledge about the composition and abundance of bacteria that are part of the microbiological community of the bovine intestine. In this case, the approach was through the analysis of feces in free grazing cattle. Although a large number of bacterial taxa were reported from the collected samples, it was not possible to determine the genus or species of some bacteria, so it is still necessary to go further into the taxonomy using specific molecular markers. However, the results obtained in the present study could be used as a bacteriological baseline for monitoring the grazing bovine intestinal health status, and to trace possible interactions with the fecal microbiota of native roaming wildlife in the area. Finally, it is important to emphasize that the next generation massive sequencing is a very effective technique that simplifies the analysis of complete bacterial communities; therefore, complementary studies on the microbiota in this and other bovine populations in Mexico are warranted.
Acknowledgments
To S.I. Barraza-Guerrero, D. Acosta-Astorga and R. Zapata-Fernández for their support in the fieldwork. The owners of the Mohovano de las Lilas locality gave their authorization to take bovine fecal samples.
Conflict of interest
The authors declare that they have no conflict of interest.
Literature cited:
- Haley BJ, Pettengill J, Gorham S, Ottesen A, Karns JS, Van-Kessel JAS. Comparison of microbial communities isolated from feces of asymptomatic Salmonella-shedding and non-Salmonella shedding dairy cows. Front Microbiol 2016;(7):691.
- Kim M, Kuehn LA, Bono JL, Berry ED, Kalchayanand N, Freetly HC, et al. The impact of the bovine faecal microbiome on Escherichia coli O157: H7 prevalence and enumeration in naturally infected cattle. J Appl Microbiol 2017;123(4):1027-1042.
- SEMARNAT-CONANP. Secretaría de Medio Ambiente y Recursos Naturales-Comisión Nacional de Áreas Naturales Protegidas). Programa de Conservación y Manejo Reserva de la Biosfera Mapimí, México. 1ª ed, México DF: Editorial EDM; 2006.
- Bell GP, Yanoff S, Karges J, Montoya JA, Najera S, Arango AM, et al. Conservation blueprint for the Chihuahuan Desert ecoregion. In: Hoyt CA, Karges J editors. Proc Sixth Symp Natur Resour Chihuahuan Desert Region. The Chihuahuan Desert Research Institute, For Davis, TX; 2014:1-36.
- Barral H. El hombre y su impacto en los ecosistemas a través del ganado. En: Montaña C editor. Estudio integrado de los recursos vegetación, suelo y agua en la Reserva de la Biosfera de Mapimí, I. Ambiente natural y humano. México, DF. Instituto de Ecología, AC. 1988:241-268.
- Álvarez-Romero J, Medellín RA. Bos taurus. En: Medellín RA, et al. editores. Vertebrados superiores exóticos en México: diversidad, distribución y efectos potenciales. Proyecto U020. Instituto de Ecología, Universidad Nacional Autónoma de México. México. D.F. Bases de datos SNIB-CONABIO; 2005.
- Clemmons BA, Martino C, Schneider LG, Lefler J, Embree MM, Myer PR. Temporal stability of the ruminal bacterial communities in beef steers. Scientific Reports 2019;9(1):1-8.
- O'Hara E, Neves AL, Song Y, Guan LL. The role of the gut microbiome in cattle production and health: driver or passenger? Ann Rev Anim Biosciences 2020;(8):99-220.
- Mackie RI. Mutualistic fermentative digestion in the gastrointestinal tract: diversity and evolution. Integrat Comparat Biol 2002;42(2):319-326.
- Madden RH, Murray KA, Gilmour A. Carriage of four bacterial pathogens by beef cattle in Northern Ireland at time of slaughter. Lett Appl Microbiol 2006;44(2):115-119.
- Febriani Y, Levallois P, Lebel G, Gingras S. Association between indicators of livestock farming intensity and hospitalization rate for acute gastroenteritis. Epidemiol Infect 2009;137(8):1073-1085.
- Callaway TR, Dowd SE, Edrington TS, Anderson RC, Krueger N, Bauer N, et al. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J Anim Sci 2010;88(12):3977-3983.
- FAS, Guide for the care and use of agricultural animals in agricultural research and teaching, 3rd ed.; Feder Anim Sci Soc. Champaing, IL, USA. 2010.
- NAM-National Academy of Medicine. Guide for the care and use of laboratory animals. Co-Produced by the National Academy of Medicine–Mexico and the Association for Assessment and Accreditation of Laboratory Animal Care International, 1st ed. Harlan: Mexico City, Mexico. 2010.
- Cornet A. Principales caractéristiques climatiques. En: Montaña C editor. Estudio integrado de los recursos vegetación, suelo y agua en la Reserva de la Biosfera de Mapimí. Publicación 23. Instituto de Ecología AC. México, DF. 1988:45-76.
- Klindworth A, Pruesse E, Schweer T, Peplies J, Quast Ch, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 2013;41(1):e1-e1.
- Illumina. 16S Metagenomic Sequencing Library Preparation, Preparing 16S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System. 2020. https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf. Accessed May 11, 2021.
- Illumina. Nextera XT DNA Library Prep Kit Reference Guide. https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/samplepreps_nextera/nextera-xt/nextera-xt-library-prep-reference-guide-15031942-05.pdf. Accessed May 11, 2021.
- García-De la Peña C, Garduño-Niño E, Vaca-Paniagua F, Díaz-Velásquez C, Barrows CW, Gomez-Gil B, et al. Comparison of the fecal bacterial microbiota composition between wild and captive bolson tortoises (Gopherus flavomarginatus). Herpetol Conserv Biol 2019;14(3):587-600.
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;(7):335.
- Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd merger. Bioinformatics 2014;(30):614-620.
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010;26 (19):2460–2461.
- Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J. Introducing EzBioCloud: a taxonomically united database of 16S rRNA and whole genome assemblies. Int J Syst Evol Microbiol 2017;(67):1613-16-17.
- Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017;5(1):27.
- Magurran A. Measuring biological diversity. Blackwell Science Ltd. Blackwell Publishing company. UK Copyright, Designs, and Patents Act 1988. 2004:100-130.
- Aricha H, Simujide H, Wang C, Zhang J, Lv W, Jimisi X, et al. Comparative analysis of fecal microbiota of grazing Mongolian cattle from different regions in Inner Mongolia, China. Animals 2021;11(7):1938.
- Li Y, Ma S, Zhang X, Huang S, Yang H, Zhao F, et al. Evaluation of bacterial and archaeal diversity in the rumen of Xiangxi yellow cattle (Bos taurus) fed Miscanthus sinensis or common mixed feedstuff. Ann microbiol 2014;64(3):1385-1394.
- O' Donnell MM, Harris HBB, Ross RP, O’Toole PW. Core fecal microbiota of domesticated herbivorous ruminant, hindgut fermenters, and monogastric animals. Microbiologyopen 2017;6(5):e00509.
- Li Y, Hu X, Yang S, Zhou J, Qi L, Sun X, et al. Comparison between the fecal bacterial microbiota of healthy and diarrheic captive musk deer. Front Microbiol 2018;(9):300.
- Li Y, Shi M, Zhang T, Hu X, Zhang B, Xu S, et al. Dynamic changes in intestinal microbiota in young forest musk deer during weaning. Peer J 2020;(8):e8923.
- Rudi K, Moen B, Sekelja M, Frisli T, Lee MR. An eight-year investigation of bovine livestock fecal microbiota. Vet Microbiol 2012;160(3-4):369-377.
- Steelman SM, Chowdhary BP, Dowd S, Suchodolski J, Janečka JE. Pyrosequencing of 16S rRNA genes in fecal samples reveals high diversity of hindgut microflora in horses and potential links to chronic laminitis. BMC Vet Res 2012;8(1):231.
- Menke S, Heurich M, Henrich M, Wilhelm K, Sommer S. Impact of winter enclosures on the gut bacterial microbiota of red deer in the Bavarian Forest National Park. Wildl Biol 2019;8(1):1-10.
- Wang X, Chen Y, Shang Y, Wu X, Wei Q, Chen J, et al. Comparison of the gut microbiome in red deer (Cervus elaphus) and fallow deer (Dama dama) by high-throughput sequencing of the V3–V4 region of the 16S rRNA gene. Sci Asia 2019;45(6):515-524.
- Zhang Z, Yang L, He Y, Luo X, Zhao S, Jia X. Composition of fecal microbiota in grazing and feedlot angus beef cattle. Animals 2021;11(11):3167.
- Deepthi M, Arvind K, Saxena R, Pulikkan J, Shamjana U, Vineet KS, Grace T. Fecal microbiota analysis exploring taxonomic diversity of hindgut microbial communities in kasaragod dwarf and Holstein Friesian cattle. 2021. Research Square, doi: https://doi.org/10.21203/rs.3.rs-732782/v1.
- Lau SKP, Teng JLL, Chiu TH, Chan E, Tsang AKL, Panagiotou G, Zhai SL, et al. Differential microbial communities of omnivorous and herbivorous cattle in southern China. Comput Struct Biotechnol J 2018;16:54-60.
- Ming L, Yi L, Siriguleng, Hasi S, He J, Hai L, et al. Comparative analysis of fecal microbial communities in cattle and Bactrian camels. PLoS One 2017;12(3):e0173062.
- Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 2016;14(1):20-32.
- Rainey FA. Orden Clostridiales, In: DeVos P, et al. editors. Bergey’s manual of systematic bacteriology. Second ed. Springer, Dordrecht, Heidelber, London, New York. Volume three. The Firmicutes 2009:736-1191.
- La Reau AJ, Meier-Kolthoff JP, Suen G. Sequence-based analysis of the genus Ruminococcus resolves its phylogeny and reveals strong host association. Microb Genom 2016:2(12).
- Rainey FA. Family V. Lachnospiraceae fam. nov. In: DeVos P, et al. editors. Bergey’s manual of systematic bacteriology. Second ed. Volume 3. The Firmicutes. London, New York: Springer, Dordrecht, Heidelber; 2009:921-968.
- Hamer HM, Jonkers D, Venema K, Vanhoutvin, S, Troost FJ, Brummer RJ. Review article: The role of butyrate on colonic function. Aliment Pharm Therapy 2008;(27): 104–119.
- Kim M, Wells JE. A meta-analysis of bacterial diversity in the feces of cattle. Curr Microbiol 2015;72(2):145-151.
- Kim M, Kim J, Kuehn LA, Bono JL, Berry ED, Kalchayanand N, et al. Investigation of bacterial diversity in the feces of cattle fed different diets. J Anim Sci 2014;92(2):683-694.
- Zhang Z, Yang L, He Y, Luo X, Zhao S, Jia X. Composition of fecal microbiota in grazing and feedlot angus beef cattle. Animals 2021;11(11):3167.
- Rodríguez J, Carcelen F, Agapito J, Barreto T, Rodríguez C, San Martín F. Identificación preliminar de microflora bacteriana en el compartimiento 1 del sistema digestivo en alpacas (Vicugna pacos). Informe Científico Tecnológico 2009;(9):63-165.
- Shepherd ML, Swecker Jr WS, Jensen RV, Ponder MA. Characterization of the fecal bacteria communities of forage-fed horses by pyrosequencing of 16S rRNA V4 gene amplicons. FEMS Microbiol Lett 2012;326(1):62-68.
- Liu X, Fan H, Ding X, Hong Z, Nei Y, Liu Z, et al. Analysis of the gut microbiota by high-throughput sequencing of the V5–V6 regions of the 16S rRNA gene in donkey. Curr Microbiol 2014;68(5):657-662.
- Grech‐Mora I, Fardeau ML, Garcia JL, Ollivier B. Sporobacter. Bergey's Manual of systematics of archaea and bacteria online. John Wiley & Sons, Inc., in association with Bergey’s Manual Trust JL. 2015:1-6.
- Durso LM, Harhay GP, Smith TP, Bono JL, DeSantis TZ, Harhay DM, et al. Animal-to-animal variation in fecal microbial diversity among beef cattle. Appl Environ Microbiol 2010;76(14):4858-4862.
- Wang L, Zhang K, Zhang C, Feng Y, Zhang X, Wang X, et al. Dynamics and stabilization of the rumen microbiome in yearling Tibetan sheep. Sci Rep 2019;9(1):1–9.
- Mamun MAA, Sandeman M, Rayment P, Brook-Carter P, Scholes E, Kasinadhuni N, et al. The composition and stability of the faecal microbiota of merino sheep. J Appl Microbiol 2019;128(1):280–291.
- Dearing MD, Kohl KD. Beyond fermentation: other important services provided to endothermic herbivores by their gut microbiota. Integr Comp Biol 2017;57(4):723-731.
- De Tarso SGDS, Oliveira D, Afonso JAB. Ruminants as part of the global food system: how evolutionary adaptations and diversity of the digestive system brought them to the future. J Dairy Vet Anim Res 2016;3(5):00094.
- Ley RE, Hamady M, Lozupone C, Tumbaugh PJ, Ramey RR, Bircher JS, et al. Evolution of mammals and their gut microbes. Science 2007;(320):1647–1651.
- Szeligowska N, Cholewińska P, Czyż K, Wojnarowski K, Janczak M. Inter and intraspecies comparison of the level of selected bacterial phyla in in cattle and sheep based on feces. BMC Vet Res 2021;17(1):1-9
- Zhu L, Wang J, Bahrndorff S. The wildlife gut microbiome and its implication for conservation biology. Front Microbiol 2021;12:1617.
- Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzãlez A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011;332:970–974.
- Zoelzer F, Burger AL, Dierkes PW. Unraveling differences in fecal microbiota stability in mammals: from high variable carnivores and consistently stable herbivores. Anim Microbiome 2021;3(1):77.
- Milani C, Alessandri G, Mancabelli L, Mangifesta M, Lugli GA, Viappiani A, et al. Multi-omics approaches to decipher the impact of diet and host physiology on the mammalian gut microbiome. Appl Environ Microbiol 2020; 86(23):e01864-20.
- Kamada N, Chen GY. Inohara N, Núñez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol 2013;14(7):685-690.
- Wolf JF, Kriss KD, MacAulay KM, Munro K, Patterson BR, Shafer AB. Gut microbiome composition predicts summer core range size in two divergent ungulates. FEMS Microbiol Ecol 2021;97(5):fiab048.
- Sahin O, Yaeger M, Wu Z, Zhang Q. Campylobacter-associated diseases in animals. Annu Rev Anim Biosci 2016;5:21-42.
- Rush JB, Edmondson MA. Infectious agents: Campylobacter. In: Hopper RM editor. Bovine reproduction. Second ed. USA: John Wiley & Sons, Inc; 2021;717-724.
- An JU, Ho H, Kim J, Kim WH, Kim J, Lee S, et al. Dairy cattle, a potential reservoir of human campylobacteriosis: epidemiological and molecular characterization of Campylobacter jejuni from cattle farms. Front Microbiol 2018;(9):3136.
- Ocejo M, Oporto B, Hurtado A. Occurrence of Campylobacter jejuni and Campylobacter coli in cattle and sheep in northern Spain and changes in antimicrobial resistance in two studies 10-years apart. Pathogens 2019;8(3):98.
- Inglis GD, Gusse JF, House KE, Shelton TG, Taboada EN. Clinically relevant Campylobacter jejuni subtypes are readily found and transmitted within the cattle production continuum but present a limited foodborne risk. Appl Enviro Microbiol 2020; 86(6): e02101-19.n.
- Elgioushy M, Rizk MA, El-Adl M, Elhadidy M, El-Khodery S. The first molecular detection of Clostridium perfringens from pneumonic cases associated with foot and mouth disease in cattle and buffalo in Egypt. Trop Anim Health Prod 2018;51(4):847-852.
- Mariano V, Nardi A, Gradassi S, De Santis P, Anniballi F, Bilei S. A severe outbreak of botulism in cattle in Central Italy. Vet Ital 2019;55(1):57-62.
- Wolf R, Hiesel J, Kuchling S, Deutz A, Kastelic J, Barkema HW, et al. Spatial-temporal cluster analysis of fatal Clostridium chauvoei cases among cattle in Styria, Austria between 1986 and 2013. Prev Vet Med 2017;138:134-138.
- Smith JS, Krull AC, Schleining JA, Derscheid RJ, Kreuder AJ. Clinical presentations and antimicrobial susceptibilities of Corynebacterium cystitidis associated with renal disease in four beef cattle. J Vet Inter Med 2020;34(5):2169-2174.
- Achollah AM, Karanja DN, Ngâ CJ, Bebora LC. Causes of organ condemnations in cattle at slaughter and associated financial losses in Siaya County, Kenya. J Vet Med Anim Health 2020;12(2):27-35.
- Kirkeby C, Halasa T, Farre M, Chehabi GN, Græsbøll K. Transmission dynamics of Corynebacterium spp. within two Danish Dairy Cattle herds. Front Vet Sci 2021;977.
- Borja E, Borja LF, Prasad R, Tunabuna T, Toribio JAL. A retrospective study on bovine tuberculosis in cattle on Fiji: Study findings and stakeholder responses. Front Vet Sci 2018;(5):270.
- Ghebremariam MK, Michel AL, Vernooij JCM, Nielen M, Rutten VP. Prevalence of bovine tuberculosis in cattle, goats, and camels of traditional livestock raising communities in Eritrea. BMC Vet Res 2018;14(1):1-13.
- Kuria JK. Diseases caused by bacteria in cattle: tuberculosis. In bacterial cattle diseases. Kaoud HA (IntechOpen) editor. 2019. London: IntechOpen; 2019. https://www.intechopen.com/chapters/64814 doi: 10.5772/intechopen.82051.
- Hacker E, Antunes CA, Mattos-Guaraldi AL, Burkovski A, Tauch A. Corynebacterium ulcerans, an emerging human pathogen. Future microbiol 2016;11(9);1191-1208.
- Amachawadi RG, Nagaraja TG. Liver abscesses in cattle: A review of incidence in Holsteins and of bacteriology and vaccine approaches to control in feedlot cattle. J Anim Sci 2016;94(4):1620-1632.
- Amachawadi RG, Tom WA, Hays MP, Fernando SC, Hardwidge PR, Nagaraja TG. Bacterial community analysis of purulent material from liver abscesses of crossbred cattle and Holstein steers fed finishing diets with or without tylosin. J Anim Sci 2021;99(4):skab076.
- Aguiar-Veloso V, Drouillard JS. On the potential role of dietary lysine as a contributing factor in development of liver abscesses in cattle. Front Vet Sci 2020;(7):733.
- Pillai DK, Amachawadi RG, Baca G, Narayanan SK, Nagaraja TG. Leukotoxin production by Fusobacterium necrophorum strains in relation to severity of liver abscesses in cattle. Anaerobe 2021;(69):102344.
- Zhivin O, Dassa B, Moraïs S, Utturkar SM, Brown SD, Henrissat B, et al. Unique organization and unprecedented diversity of the Bacteroides (Pseudobacteroides) cellulosolvens cellulosome system. Biotechnol Biofuels 2017;10(1):1-19.
- Duncan JS, Leatherbarrow AJH, French NP, Grove-White DH. Temporal and farm-management-associated variation in faecal-pat prevalence of Campylobacter fetus in sheep and cattle. Epidemiol Infect 2014;142(6):1196-1204.
- Siembieda JL, Kock RA, McCracken TA, Newman SH. The role of wildlife in transboundary animal diseases. Anim Health Res Rev 2011;12(1):95-111.
- Cossío-Bayúgar A, Romero E, Gallina S, Suzán G, Ibáñez-Bernal S. Variation of gastrointestinal parasites in mule deer and cattle in Mapimí biosphere reserve, Mexico. Southwest Nat 2015;60(2-3):180-185.
- McAllister TA. Topp E. Role of livestock in microbiological contamination of water: commonly the blame, but not always the source. Anim Front 2012;2(2):17-27.
- Gortázar C, Ferroglio E, Höfle U, Frölich K, Vicente J. Diseases shared between wildlife and livestock: A European perspective. Eur J Wildl Res 2007;53(4):241-256.
- Espunyes J, Cabezón O, Dias-Alves A, Miralles P, Ayats T, Cerdà-Cuéllar M. Assessing the role of livestock and sympatric wild ruminants in spreading antimicrobial resistant Campylobacter and Salmonella in alpine ecosystems. BMC Vet Res 2021;17(1):1-8.