https://doi.org/10.22319/rmcp.v13i3.5985
Revisión bibliográfica
La citometría de flujo, un universo de posibilidades en el ámbito veterinario. Revisión
Luvia Enid Sánchez-Torres a*
Alejandra Espinosa-Bonilla b
Fernando Diosdado-Vargas c
a Instituto Politécnico Nacional. Escuela Nacional de Ciencias Biológicas, Departamento de Inmunología. Prolongación de Carpio y Plan de Ayala s/n, Colonia Santo Tomás, Alcaldía Miguel Hidalgo, 11340, Ciudad de México, México.
b Instituto Politécnico Nacional. Escuela Nacional de Ciencias Biológicas, Central de Instrumentación. México.
c Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias. Ciudad de México, México.
* Autor de correspondencia: luviasanchez@hotmail.com
Resumen:
La citometría de flujo es una tecnología que ha favorecido el rápido avance de muchas y muy diversas áreas de la ciencia, ya que permite la medición simultánea de múltiples características de cada una de las partículas o células individuales que se encuentran en una muestra mientras pasan a gran velocidad a través de una zona iluminada por uno o varios láseres. La información obtenida incluye datos sobre el tamaño y la complejidad interna, así como otros parámetros inherentes a cada una de las partículas presentes en la muestra, los cuales son captados por el equipo como señales luminosas. Las partículas más comúnmente analizadas en los citómetros de flujo son células, así que puede analizarse la expresión de moléculas tanto en su superficie como en su interior, la viabilidad, la funcionalidad, la proliferación celular, el contenido de ADN y la producción de citocinas entre muchas otras. Estas determinaciones pueden realizarse gracias a la utilización de anticuerpos acoplados a fluorocromos o bien, al uso de moléculas cuya fluorescencia depende de la característica que se quiera evaluar. Algunos citómetros de flujo son además clasificadores (“sorters”), lo que implica que el equipo puede separar físicamente las células que presentan las características de interés y además, es factible que una vez que han sido purificadas, éstas puedan emplearse en posteriores experimentos. En esta revisión se concentran los fundamentos de la citometría de flujo y sus principales aplicaciones, las cuales ofrecen una gran ventana de oportunidad en el ámbito veterinario, tanto en investigación como en la clínica.
Palabras clave: Citometría de flujo, Inmunofenotipo, Viabilidad, Muerte celular, Análisis de DNA, Citocinas.
Recibido: 28/04/2021
Aceptado: 17/11/2021
Generalidades de la citometría de flujo
La citometría de flujo (CF) permite analizar simultáneamente varias características individuales de células o partículas en suspensión a medida que pasan por uno o varios haces de luz láser. Los citómetros de flujo pueden leer miles de células por segundo con la posibilidad de regular la velocidad de análisis, de tal manera que el análisis multiparamétrico y la velocidad a la que se realiza, constituyen dos de sus principales y más poderosas ventajas. El tipo de muestra que puede utilizarse es muy diverso e incluye sangre, poblaciones celulares purificadas, líneas celulares, suspensiones celulares de órganos sólidos, núcleos extraídos de bloques de parafina, organelos celulares, liposomas, vesículas extracelulares y líquidos corporales, entre otros. Las células que pueden estudiarse en un citómetro de flujo pueden provenir de diferentes especies animales, vegetales e incluso, es posible hacer estudios directamente en microorganismos(1-5). Debido a la posibilidad de estudiar también partículas inertes, es factible analizar y cuantificar moléculas en solución, las cuales pueden estar en muestras de suero, plasma, orina, líquido cefalorraquídeo, calostro, semen, sobrenadantes de cultivos, etc.(6,7).
Dada su gran versatilidad, la CF es actualmente una de las técnicas más utilizadas en diversas áreas. La relevancia de sus aportaciones y la transcendencia de los resultados obtenidos ha llevado a integrar asociaciones nacionales e internacionales que han permitido compartir información y establecer protocolos unificados (Páneles de inmunofluorescencia multicolor optimizados, “OMIP” por sus siglas en inglés) para diversas aplicaciones, principalmente aquellas relacionadas con la caracterización de poblaciones celulares y con el diagnóstico clínico, algunas de las cuales están enfocadas al campo veterinario(8-11), así como revistas especializadas en el tema como “Cytometry”(12). El uso de esta tecnología en el ámbito veterinario se ha ido incrementado de manera lenta y gradual; se tienen reportadas múltiples aplicaciones no solo en investigación sino en la clínica, tanto para animales de compañía como para animales de vida salvaje. Los aspectos de la medicina veterinaria que más se han beneficiado de la CF son el diagnóstico, el pronóstico y la inseminación artificial(13-19); desafortunadamente en México, su uso en la clínica veterinaria es escaso ya que no hay laboratorios que cuenten con alguno de estos equipos. En el área de investigación veterinaria, diversas instituciones educativas cuentan con equipos en los cuales se pueden realizar ese tipo de análisis, siendo aún poco el uso que se hace de esta tecnología. Es por lo anterior que, en este documento se pretende recalcar su versatilidad y dar a conocer las múltiples áreas de oportunidad que existen en el ámbito veterinario para aprovechar todas las aplicaciones de la citometría de flujo, sobre todo en la clínica.
Fundamentos de la citometría de flujo
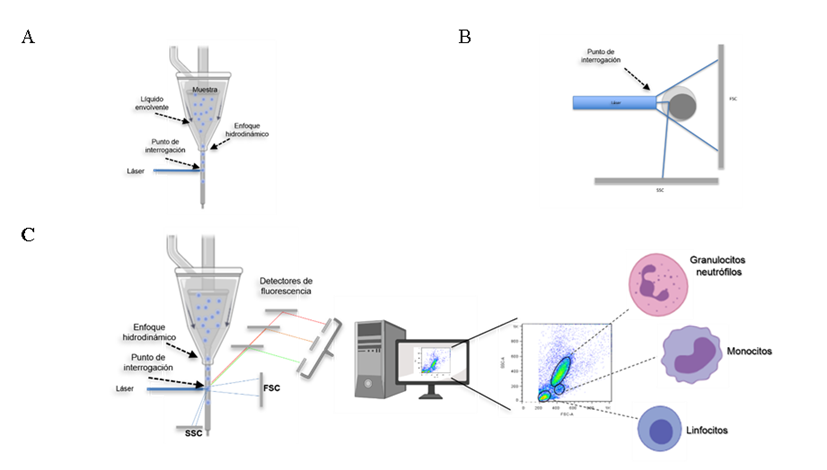
Las propiedades que pueden ser medidas por un citómetro de flujo incluyen el tamaño, la complejidad interna y la intensidad de fluorescencia de las células analizadas. Todos estos parámetros son determinados de manera relativa y no se generan valores absolutos, a menos que se utilicen estándares y controles de referencia(20,21). Las partículas o células que van a ser analizadas deben estar en suspensión, de tal manera que son tomadas por el equipo y son dirigidas a un espacio físico llamado por algunos autores como “punto de interrogación”, que es el sitio en el cual, el láser o láseres del equipo inciden sobre las células (Figura 1A). Cuando el láser incide en cada una de las células de la muestra, provoca la dispersión de la luz en varias direcciones, lo cual da información sobre su tamaño y complejidad relativas (Figura 1B); si además hay moléculas fluorescentes presentes en la célula, el equipo capta la fluorescencia emitida por estas, pudiendo dar información sobre la expresión de moléculas y algunas funciones celulares, entre otras características como se detalla más adelante (Figura 1C). La magnitud de cada una de las señales para tamaño, complejidad y fluorescencia es registrada para todas y cada una de las células de la muestra que pasan a través del láser, por lo que es una tecnología que hace un análisis individual, célula por célula. Finalmente, las señales emitidas son recolectadas y transformadas en valores que pueden ser analizados por una computadora e interpretadas por los usuarios de manera sencilla (Figura 1C)(22).
Figura 1: Funcionamiento general de un citómetro de flujo
La muestra es llevada al sitio físico en el cual el láser incide sobre cada una de las células llamado punto de interrogación. El alineamiento de las células se logra gracias a la diferencia de presión a la cual circula la solución que va por fuera de la muestra (líquido envolvente), generando el enfoque hidrodinámico de las células (A). Cuando el láser incide sobre cada célula es desviado con base en el tamaño y la complejidad interna de cada una de ellas; dichas señales son detectadas en el detector frontal (FSC) y el detector lateral (SSC) respectivamente (B). Los detectores de tamaño, complejidad y los que captan las señales fluorescentes, llevan la información a una computadora para que ésta la muestre en gráficos sencillos de interpretar (C). Imagen creada parcialmente con BioRender.
Componentes de un citómetro de flujo
Los citómetros de flujo están compuestos de tres sistemas principales: sistema de fluidos, sistema óptico y sistema electrónico(2,23).
El sistema de fluidos toma la muestra y dirige a las células al punto de interrogación. Para que las partículas sean iluminadas de la mejor manera, éstas deben pasar una por una por el centro del haz del rayo láser. Lo anterior se logra gracias al enfoque hidrodinámico, que favorece el alineamiento de las células en el flujo gracias a la diferencia de presión que hay entre la suspensión celular y el líquido que va por la parte externa, llamado líquido envolvente, como se muestra en la figura 1A(24,25).
El sistema óptico está compuesto por dos sistemas, el de excitación (láseres) y el colector de señales(26). Cuando el láser se encuentra con una célula, éste se dispersa dependiendo de las propiedades físicas de la célula, en particular de su tamaño y su complejidad interna. La luz dispersada es captada por un detector frontal (forward scatter = FSC) y el valor reportado es proporcional a la superficie celular o tamaño de la partícula iluminada. Por otro lado, la luz que se dispersa lateralmente es captada por otro detector ubicado a 90° del láser (side scatter = SSC), en este caso, el valor generado es proporcional a la complejidad interna de la célula o de la partícula(22-23). En la figura 1B, se muestra un esquema representativo. Con base en los valores de FSC y el SSC de cada célula en la muestra, el sistema electrónico de los citómetros construye gráficos que permiten ubicar en posiciones diferentes, a aquellas células de la muestra que tengan diferencias suficientes entre ellas en cuanto a su tamaño o complejidad. Es decir, se toma una muestra de sangre periférica, se lisan sus eritrocitos y se pasan por un citómetro, los linfocitos aparecerán en el gráfico en una posición diferente a la de los neutrófilos, ya que los primeros son pequeños y su complejidad interna es baja, mientras que los neutrófilos son más grandes y son más complejos en su interior dado que su núcleo es multilobulado y tiene gran cantidad de gránulos (Figura 1C)(1-2). Cabe mencionar que la ubicación en el gráfico de FSC y SSC no da la identificación de las poblaciones celulares; como se verá más adelante, se requiere del uso de anticuerpos para definir sin lugar a duda, la identidad de las células presentes y la proporción en la que se encuentra cada una en la muestra que se analiza.
El sistema electrónico se encarga de hacer la conversión de las señales ópticas en señales electrónicas proporcionales o pulsos de voltaje. Las señales de luz son generadas conforme cada una de las células pasa por el haz del láser. Estas señales de luz son transformadas en señales electrónicas mediante fotodetectores y con base en su intensidad, se les asigna un valor relativo en una escala. Las señales con idénticas intensidades se acumulan en el mismo valor de la escala, lo que incrementa la altura del pico y las señales de mayor intensidad se representan en los valores más altos de la escala del gráfico. Finalmente, todos estos valores se presentan de manera en que los usuarios pueden interpretar los resultados obtenidos por el equipo, como se ejemplifica más adelante(22,25,27).
Aunado a la información que se puede tener de las células o partículas con respecto a su tamaño y complejidad interna, se puede obtener información sobre otras características basada en señales fluorescentes(28). Un compuesto fluorescente o fluorocromo es capaz de absorber la luz dentro de un rango determinado de longitudes de onda y en consecuencia de emitir a una longitud de onda mayor a la de absorción. El rango de longitudes de onda en las cuales un compuesto fluorescente puede excitarse es llamado espectro de absorción, y el rango de longitudes de onda de los fotones emitidos es denominado espectro de emisión(25).
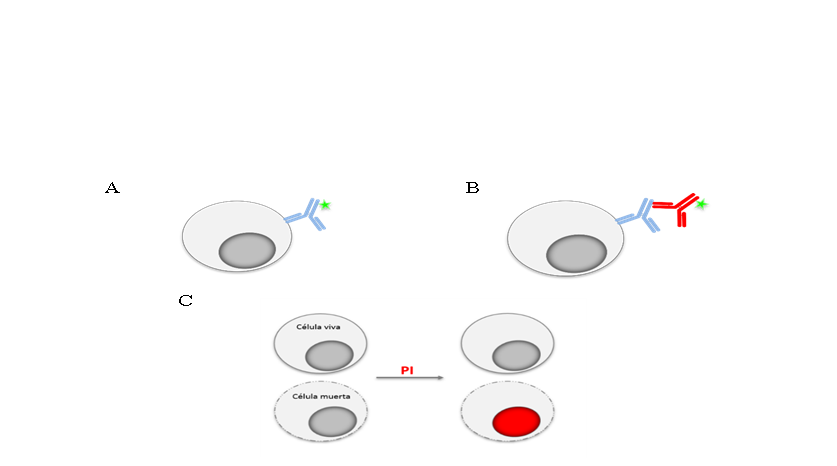
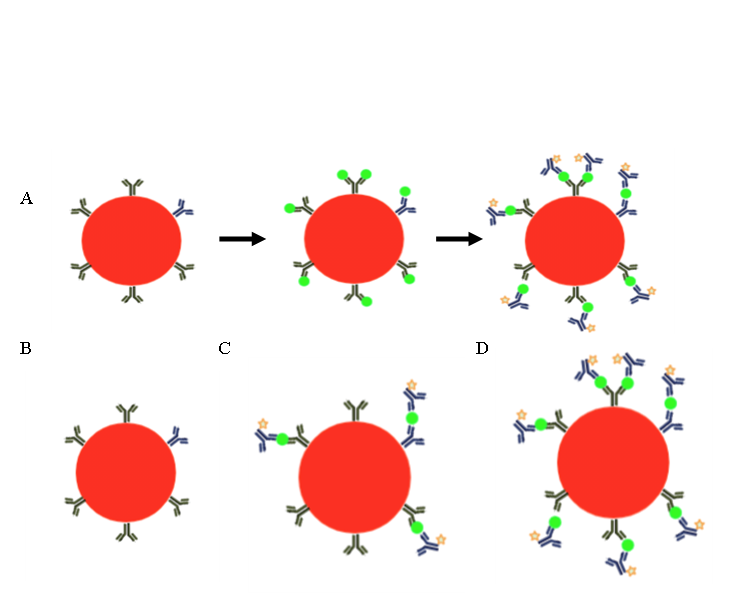
La fluorescencia que puede ser detectada en un citómetro puede ser intrínseca o extrínseca, es decir, puede provenir de moléculas que forman parte de la célula (riboflavinas, NADPH, triptófano, tirosina, etc.) y por ello suelen denominarse como autofluorescencia, o bien provenir de algún reactivo fluorescente que se añadió a la muestra(1). Los reactivos más utilizados en CF son los anticuerpos, los cuales se unen a la molécula contra la cual fueron fabricados y que se quiere detectar. Estos anticuerpos, llamados anticuerpos primarios, pueden estar marcados con algún fluorocromo (Figura 2A) o bien, requerir de un segundo anticuerpo o anticuerpo secundario que reconozca al anticuerpo primario y que sea el secundario el que esté conjugado al fluorocromo (Figura 2B)(23). En algunas de las aplicaciones de la citometría no se utilizan anticuerpos como herramientas sino moléculas cuya longitud de onda de emisión o bien la intensidad de fluorescencia, depende de la característica celular que se quiere evaluar (Figura 2C).
Figura 2: Tinción para citometría de flujo con anticuerpos y moléculas fluorescentes
Para analizar la expresión de moléculas en las células se pueden utilizar anticuerpos específicos para ellas, los cuales pueden ir unidos a un fluorocromo (estrella verde) (A) o necesitar un segundo anticuerpo acoplado a un fluorocromo que reconoce al anticuerpo primario (B). Para algunas aplicaciones se utilizan moléculas cuya fluorescencia indica alguna característica o función de la célula. En (C) se ejemplifica el uso de ioduro de propidio (PI), molécula fluorescente que se añade a la muestra; las células vivas no dejan pasar el fluorocromo a su interior por lo que no fluorescen, mientras que las muertas sí, ya que tienen su membrana dañada, dejando pasar el PI y tiñendo los ácidos nucleicos de la célula.
La CF actual se considera multiparamétrica ya que es posible evaluar además de tamaño y complejidad, varias características fenotípicas y funcionales en una misma célula de manera simultánea(22,29). El número de fluorocromos y, en consecuencia, el número de características que se pueden analizar en una misma muestra, depende del modelo y la configuración del citómetro en el que se trabaje, así como de las características propias de cada fluorocromo que se quiera utilizar.
El láser más comúnmente usado en CF es el de argón, el cual tiene una longitud de onda de 488 nm (azul). La mayoría de los citómetros de flujo actuales tienen más de un láser, lo que incrementa su potencial de análisis y su versatilidad(26). Los fluorocromos que pueden utilizarse en un citómetro son aquellos que pueden ser excitados por alguno de los láseres del equipo y que pueden ser captados por los detectores de fluorescencia que tiene. Las longitudes de onda que capta cada detector de fluorescencia están definidas por los filtros que tenga el equipo. Algunas aplicaciones que facilitan la selección de fluorocromos con base en la configuración del equipo disponible son BD Bioscience Spectrum Viewer, Biolegend Spectra Analyzer, FluoroFinder y ThermoFisher Fluorescence Spectra Viewer.
Tipos de gráficos y su interpretación
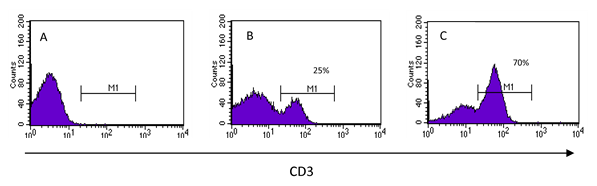
Los datos que proporcionan los citómetros de flujo de todas y cada una de las células de una muestra, deben presentarse de manera que su interpretación sea fácil, rápida e integrativa. Los histogramas se emplean para analizar uno solo de los parámetros captados por el equipo, el cual se visualiza en el eje “x”, mientras que en el “y” se representa el número de eventos, ya sean células o partículas que cumplen con esa característica; los histogramas son en realidad gráficos de frecuencias (Figura 3). Con este tipo de gráfico se puede distinguir entre eventos que presentan o no la característica evaluada en el eje “x”. La interpretación más sencilla de este tipo de gráficos es de todo o nada, es decir, o tiene o no tiene la molécula que se quiere identificar, pero la utilidad de este tipo de gráfico va más allá, ya que permite saber qué tanto la expresa y poder comparar niveles de expresión de una molécula entre las células de una misma muestra o entre muestras que se encuentren en condiciones diferentes. Para poder establecer el porcentaje de células positivas para la marca, debe darse al citómetro una muestra a la que no se agregó la marca fluorescente para que sirva de punto de referencia entre lo negativo y lo positivo. En la figura 3 se muestra un ejemplo en el que se evalúa la expresión de la molécula CD3 en las células de una muestra. En la figura 3A, se muestra el histograma de una muestra no teñida, lo que implica que, cuando se lea una muestra teñida, todas las células que aparezcan a la derecha (en los valores de la escala que comprende M1, figuras 3B y3C) serán positivas para la marca que se esté determinando, siendo posible obtener el porcentaje de éstas(1,23,30).
Figura 3: Análisis unidimensional
En los histogramas se visualiza una sola característica a la vez. Para saber si las células expresan la molécula de interés (CD3), primero se introduce una muestra que no haya sido teñida (A) y con ella se establece a partir de qué valor en la escala se considerará que las células son positivas (M1). En (B) y (C) se muestran histogramas de dos muestras diferentes teñidas en las que hay 25 % y 70 % de células que expresan la molécula CD3 respectivamente.
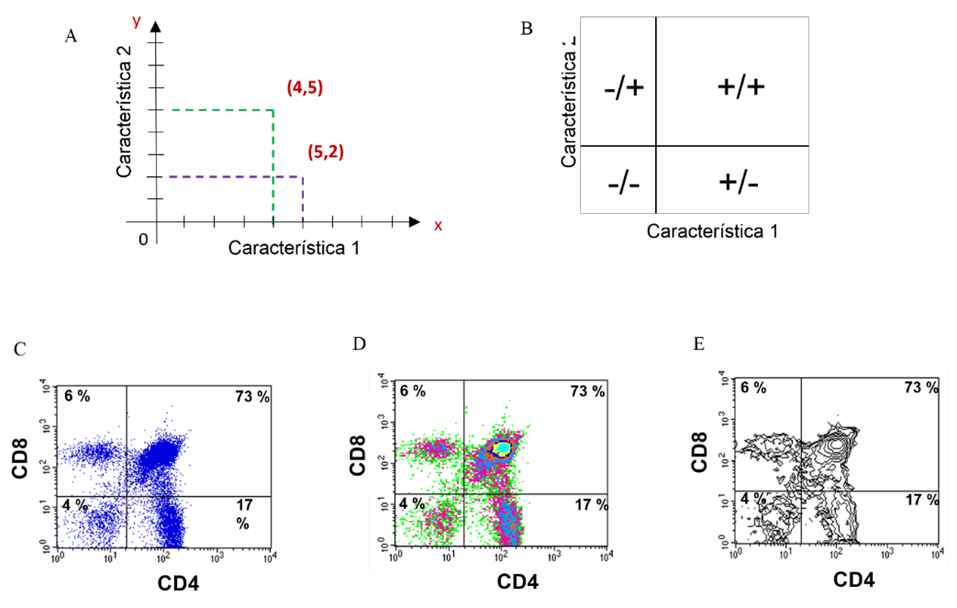
Los diagramas de puntos, de densidad o de contornos son ejemplos de gráficos que permiten correlacionar dos parámetros al mismo tiempo, uno de ellos graficado en el eje “x” y el otro en el eje “y”. La diferencia entre estos gráficos es la manera en que presenta dicha interacción. La ubicación de cada célula en el gráfico es similar a lo que ocurre con coordenadas en un plano cartesiano, en el que la posición de cada célula dependerá de sus valores individuales para la característica graficada en x y para la graficada en y (Figura 4A). Con este tipo de representación, se pueden distinguir al menos cuatro posibilidades: las células que no presentan ninguna de las dos características que se evalúan y que se ubican en el cuadrante inferior izquierdo, las que solo presentan una u otra y por ello se visualizarán en los cuadrantes superior izquierdo o inferior derecho, y aquellas que presentan ambas y por lo tanto se ubican en el cuadrante superior derecho (Figura 4B). Cuando se realiza el análisis de los resultados, el citómetro de flujo reportará el porcentaje de células que tienen la o las características de mayor interés(23,30). En las figuras 4C-E se muestra el análisis biparamétrico de una suspensión de timo teñido con un anticuerpo anti-CD4-FITC (característica 1) y un anticuerpo anti-CD8-PE (característica 2). Se observa que en la muestra existe un 4 % de células que no expresan la molécula CD4, ni la molécula CD8, un 17 % que solo expresan CD4, un 6 % que solo expresan CD8 y un 73 % de células que expresan tanto la molécula CD4 como la CD8. La visualización de la expresión y coexpresión de ambas moléculas puede hacerse en un diagrama de puntos (dot plots, figura 4C), un diagrama de densidad (density plots, figura 4D) o en un diagrama de contornos (contour plots, figura 4E).
Figura 4: Análisis bidimensional
Se visualizan dos características de manera simultánea y se pueden establecer correlaciones entre ellas. Cada célula (punto) tiene un valor en el eje x y otro en y, similar a un plano cartesiano (A); Interpretación general para diagramas bidimensionales (B); Ejemplos de gráficos bidimensionales: Diagrama de puntos (C), diagrama de densidades (D) y diagrama de contornos (E) de la misma muestra.
Sin duda, una de las áreas de mayor avance en los últimos años dentro de la CF tiene que ver con el desarrollo de softwares cada vez más robustos, los cuales hacen uso de la minería de datos y el machine learning que permiten hacer análisis multivariados complejos. Lo anterior ha permitido que ahora sea muy frecuente encontrar los resultados de CF organizados en mapas de calor o en análisis de componentes principales entre otras muchas estrategias, las cuales permiten analizar muchos parámetros integrados en un solo gráfico y comparar los resultados entre individuos o condiciones. Los archivos que se generan en los citómetros de flujo utilizan el formato de archivo cuya extensión es “.fcs” (flow cytometry standard), lo cual facilita el análisis de los archivos generados en diferentes plataformas, independientemente del citómetro de flujo en el que hayan sido adquiridas las muestras(22,31,32).
Separación celular (“cell sorting”)
Algunos citómetros de flujo tienen la capacidad de hacer “sorting”, es decir, de enriquecer una población particular, la cual es separada físicamente del resto de las células de la muestra; al final, se obtiene la o las poblaciones de interés purificadas o enriquecidas de manera aséptica en tubos individuales o en pozos de placas de cultivos, con la opción de realizar estudios posteriores con las células así obtenidas, como análisis por microscopia, cultivo celular y biología molecular entre otros(33,34).
Aplicaciones clásicas de la citometría de flujo
Como ya se ha mencionado, la CF es una tecnología extremadamente versátil que evalúa varios parámetros de manera simultánea en cada célula, lo que permite establecer correlaciones entre ellos. Con la CF no solo se pueden analizar la presencia de moléculas en la superficie o en el interior de las células, sino también cuantificarlas. Es posible hacer pruebas funcionales, evaluaciones biológicas de compuestos tanto en células eucariotas como procariotas, purificar poblaciones celulares aun cuando se encuentren en baja proporción en la muestra, cuantificar moléculas solubles y evaluar la proliferación celular, la viabilidad y el tipo de muerte celular que presenta una célula, la actividad metabólica y la señalización intracelular solo por mencionar algunas; otro aspecto muy importante es que al poderse evaluar de manera simultánea muchas de estas características, se reduce el volumen de muestra necesario(35,36). Las técnicas que se describen a continuación tienen aplicación tanto en humanos como en el ámbito veterinario, y dentro de éste, tanto en animales domésticos como en fauna silvestre. Cabe mencionar que, en algunas ocasiones, la limitante es la disposición en el mercado de los anticuerpos que reconocen moléculas específicas de las diferentes especies animales, pero dada la importancia de los resultados que pueden obtenerse por CF, la disponibilidad de anticuerpos con diferentes especificidades ha crecido en los últimos años, facilitando el trabajo en el área pecuaria tanto en investigación como en la clínica, sobre todo para diagnóstico. Aunado a lo anterior, se ha reportado reactividad inter-especies de diferentes anticuerpos, lo que permite su uso aún en aquella para la cual no fueron producidos originalmente(37).
Inmunofenotipo
El inmunofenotipificación hace referencia a la caracterización de las poblaciones celulares con base en las moléculas que expresa una célula, permitiendo así su identificación gracias al uso de anticuerpos(28). Para realizar un estudio de inmunofenotipificación se utilizan anticuerpos que reconocen moléculas particulares en las células y con base en su presencia o ausencia, o a su nivel de expresión, es que se realiza dicha identificación o caracterización. Hay que recordar que existen moléculas que se expresan en varias estirpes celulares, mientras que la expresión de otras es exclusiva de ciertas poblaciones o subpoblaciones celulares, lo que implica que debe hacerse una correcta selección de anticuerpos. Debe tenerse en cuenta que las moléculas que identifican a cada población celular pueden depender de la especie animal con la que se esté trabajando(38-42).
La detección de las diferentes moléculas de interés se realiza utilizando anticuerpos marcados con fluorocromos, por lo que si se hace incidir un haz de luz que excite a ese fluorocromo, éste emitirá fluorescencia a cierta longitud de onda que será captada por un detector que identificará al fluororomo y en consecuencia a la molécula a la que se unió el anticuerpo. La identificación o caracterización de las poblaciones celulares puede ser tan detallada como se necesite. Algunos citómetros como se mencionó previamente, sólo permiten evaluar de manera simultánea 1-4 parámetros basados en fluorescencia, es decir solo se pueden emplear un máximo de cuatro anticuerpos que reconocen moléculas diferentes, mientras que en otros equipos, se puede estudiar la expresión de más de 20 moléculas en un mismo tubo, lo cual amplía las posibilidades y el detalle de la caracterización y cuantificación de las poblaciones celulares(28). Las figuras 4C-E son ejemplos de inmunofenotipificación de diferentes muestras utilizando dos anticuerpos, un anti-CD4 y un anti-CD8.
La inmunofenotipificación ha resultado de vital importancia en el diagnóstico y pronóstico de diversas enfermedades veterinarias y un complemento muy adecuado al estudio morfológico convencional, dado que es factible detectar modificaciones en las proporciones de las diferentes poblaciones celulares, así como caracterizar células neoplásicas con base en sus marcadores fenotípicos(43), lo cual permite hacer un diagnóstico certero. Actualmente, uno de los principales usos de la inmunofenotipificación ha sido en la detección y caracterización de procesos hemato-oncológicos, tanto linfo como mieloproliferativos en pequeñas especies(13,14,16,44). A manera de ejemplo, se puede mencionar el caso de los linfomas caninos, los cuales son el tipo de tumor hematológico más frecuente; del 30 al 40 % son linfomas de células T y el resto son de células B, los cuales pueden diferenciarse con base en los marcadores fenotípicos que presentan. Los paneles de anticuerpos utilizados pueden llevar a una caracterización más fina que permite, de ser necesario, identificar incluso diferentes subtipos. La información sobre los anticuerpos, los marcadores fenotípicos de cada población y subpoblación celular, así como la estrategia de análisis pueden ser consultados en diferentes trabajos publicados recientemente(45), en los cuales se menciona que la FC puede utilizarse además, para dar seguimiento al tratamiento y detectar enfermedad mínima residual en estos animales, lo que puede ser indicio de una recaída. Gracias a la CF se han identificado ciertos fenotipos con un peor pronóstico(45). Es interesante ver que algunos anticuerpos obtenidos para humanos muestran reactividad cruzada con antígenos caninos, lo que permite su utilización. La rapidez con la que se pueden obtener el diagnóstico es una ventaja adicional.
Por otro lado, en los hatos lecheros, la cuantificación de células somáticas en la leche es una estrategia común que permite detectar la presencia de infecciones clínicas y subclínicas en las vacas. La CF permite hacer una cuenta diferencial de poblaciones y subpoblaciones celulares presentes en la leche, lo que ha sido sugerido como una excelente alternativa para identificar procesos inflamatorios en las ubres, aun cuando las cuentas de células somáticas son bajas. La detección temprana de procesos infecciosos, evita su progresión y con ello, cambios en la cantidad y calidad de la leche, reduciendo las posibles pérdidas económicas(40,46).
Viabilidad celular
El determinar la viabilidad de las células de una muestra permite valorar el estado en el que se encuentran, factor que puede constituir un control de calidad interno dentro del laboratorio(47,48). La viabilidad de las células de una muestra depende de muchos factores, entre ellos se pueden citar el procedimiento de obtención, el transporte (si éste es necesario), el procesamiento de la muestra durante la tinción, el almacenamiento, etc., de tal manera que, si la muestra presenta una viabilidad baja, habrá que determinar cuál o cuáles factores están afectando este parámetro biológico. Es importante mencionar que la presencia de células muertas en una muestra, favorece la unión de los anticuerpos de manera independiente a la especificidad de estos, por lo que, si no se eliminan las células muertas del análisis, éstas pueden llevar a resultados y conclusiones erróneas debido a la presencia de eventos falsos positivos(48,49). Por otro lado, es posible que haya una disminución de la viabilidad (y por ende, un incremento de la muerte celular) como consecuencia de una infección, de un tratamiento con quimioterapéuticos, de una estimulación celular in vitro, etc., evento que debe llamar la atención y requerir un estudio detallado de las causas e implicaciones. En el caso particular de los quimioterapéuticos, el incremento en el porcentaje de células muertas puede indicar una buena respuesta al tratamiento.
Los reactivos para cuantificar el porcentaje de viabilidad (o de muerte celular) o para excluir a las células muertas de un análisis de CF se dividen en dos grandes grupos. En el primer caso, el fundamento implica que dada que las células muertas tienen dañada la membrana celular, el fluorocromo entra y tiñe la célula muerta, mientras que las células vivas no captan el fluorocromo porque su membrana está íntegra, pudiendo distinguirlas como teñidas y no teñidas respectivamente (Figura 2C). Ejemplos de este tipo de moléculas son el ioduro de propidio (PI) y el 7-amino, actinomicina D (7-AAD); las células una vez teñidas no se pueden fijar. En el segundo caso, una vez agregado el reactivo, éste se lava y las células se fijan, lo que permite que la lectura en el citómetro no tenga que realizarse inmediatamente y las muestras puedan almacenarse. En este caso, las moléculas fluorescentes que se utilizan se unen covalentemente a las proteínas de las células. En las células vivas, al tener la membrana intacta, las únicas proteínas que reaccionarán con el reactivo serán las de la superficie. En el caso de las células muertas, el reactivo además reaccionará con las proteínas del interior de la célula quedando más intensamente teñidas que las células vivas, haciendo posible su distinción(47,50). Los gráficos que se obtienen en ambos casos y su interpretación son similares independientemente del tipo de reactivo que se haya utilizado. La determinación de la viabilidad de los espermatozoides antes y después del proceso de criopreservación es un ejemplo del uso rutinario de esta aplicación de la CF y constituye una evaluación indispensable en inseminación artificial en diferentes especies animales(51,52).
Muerte celular
El estudio de la muerte celular y de los posibles mecanismos que la pueden inducir ha sido un tema de gran interés. Desde la descripción de la apoptosis y posteriormente de todos los demás mecanismos descritos a la fecha, las técnicas para evidenciarla, cuantificarla y caracterizarla se han incrementado año con año, dado el mayor conocimiento sobre las vías de señalización que se activan y que llevan a la muerte de las células(49,53,54).
La muerte celular puede estudiarse de manera general desde dos perspectivas. La primera implica el establecer el porcentaje de células muertas en una suspensión celular, así como para establecer el efecto de diferentes estímulos sin que interese el mecanismo de muerte. Este tipo de determinaciones son muy comunes cuando se hacen estudios de actividad biológica de nuevas moléculas con posible actividad antibiótica o antineoplásica. Para lo anterior, se realizan tinciones similares a las descritas para viabilidad, pero lo que se reporta es el porcentaje de células muertas en la muestra analizada y como ya se mencionó, los estudios pueden hacerse no solo en células eucariotas infectadas, sino en microorganismos directamente, lo que resulta muy útil para los estudios de resistencia a antibióticos y la evaluación de nuevas moléculas antimicrobianas(55,56).
Cuando se requiere identificar el mecanismo de muerte celular es importante establecer una estrategia que permita separar los posibles eventos biológicos implicados en la muerte de las células, como la expresión de moléculas específicas de cada mecanismo de muerte, la activación de enzimas, la alteración de funciones celulares como el potencial de membrana mitocondrial y la producción de especies reactivas de oxígeno, el reacomodo de fosfolípidos en la membrana, la fragmentación del ADN, etc., Estos eventos guardan relación con los diferentes tipos de muerte celular hasta ahora descritos y con las vías se señalización que se activan en cada uno(53,57). A la fecha se conocen más de diez mecanismos diferentes de muerte celular, algunos de ellos están interconectados. Uno de los usos más frecuentes del análisis de muerte celular en al ámbito veterinario es completar el estudio de la calidad de semen en diferentes especies animales(52).
Pruebas funcionales
La CF permite también evaluar la capacidad funcional de las células; la detección de alteraciones en las capacidades normales de las células puede ser indicio de patologías y su identificación puede ayudar al diagnóstico. A continuación, se mencionan algunas de las técnicas más utilizadas y reportadas en la literatura.
Fagocitosis. La fagocitosis forma parte de los mecanismos de respuesta inmune innata que permiten la contención de infecciones por lo que alteraciones en alguno de los pasos de este proceso repercute en la salud de los animales. Los neutrófilos son las células fagocíticas más abundantes en la circulación de muchos mamíferos, aunque su proporción puede variar; en la mayoría de los carnívoros y en los caballos representan más del 50 % de las células en sangre, en los cerdos están en un 50 %, mientras que en los roedores y los rumiantes, están en un 25 % en promedio; En reptiles, aves, conejos y peces, las células fagocíticas se denominan heterófilos y su porcentaje es variable entre ellos(58).
La CF permite el estudio de la fagocitosis desde varios puntos de vista; el más común es midiendo la capacidad de fagocitar partículas o bien, evidenciando los cambios bioquímicos intracelulares que se presentas en las células después de fagocitar y que llevan a la destrucción intracelular de los microorganismos. En el primer caso, se pueden utilizar bioindicadores como bacterias o levaduras, o bien partículas inertes acopladas a un fluorocromo, por lo que se puede determinar el porcentaje de células que fagocitan y el nivel de fagocitosis(59). Además, es factible monitorear el cambio de pH una vez que han fagocitado los bioindicadores o las partículas inertes. Para esto se usan sondas fluorescentes sensibles a los cambios de pH, lo que permite monitorear cada etapa del proceso de fagocitosis, desde la formación del fagosomas hasta la fusión de estos con los lisosomas (fagolisosomas), dadas las diferencias en el pH de ambos compartimentos (pH 6.7 y 4.7 respectivamente)(60,61). Este tipo de análisis permite detectar alteraciones en la fagocitosis, las cuales se han reportado en enfermedades veterinarias como la anaplasmosis, clamydiosis, parvovirus canino, la deficiencia de adhesión leucocitaria y enfermedad granulomatosa crónica entre otras(58,62).
Activación celular. La activación de las células es el resultado de su interacción con agentes extraños o con mitógenos y puede darse in vivo o in vitro; en algunos casos la activación se manifiesta con la expresión de novo o el incremento de la expresión basal de moléculas características y puede ir o no, acompañada de proliferación celular y de síntesis de moléculas solubles como citocinas y quimiocinas que se liberan al microambiente, entre otros eventos. El detectar células activadas en un organismo implica que se está montando una respuesta inmunológica ya sea contra un microorganismo o contra un agente vacunal y junto con la inmunofenotipificación, puede dar información importante en casos de enfermedades infecciosas.
Proliferación. La proliferación celular puede evaluarse in vitro e in vivo, agregando análogos de nucléotidos como la bromodesoxiuridina (BrdU) para que, si hay proliferación y por lo tanto síntesis de ADN, esta molécula se incorpore en las nuevas cadenas de ácidos nucleicos. Para determinar si se incorporó, se utilizan anticuerpos dirigidos contra la BrdU marcados con fluorocromos, de tal manera que si las células proliferaron, estas darán señal fluorescente y se podrá cuantificar el porcentaje de células que se dividieron(63). Cabe mencionar que la BrdU tiene otros usos como evidenciar la fragmentación del ADN, por lo que no debe confundirse los fundamentos de su uso en cada caso y la interpretación de los resultados.
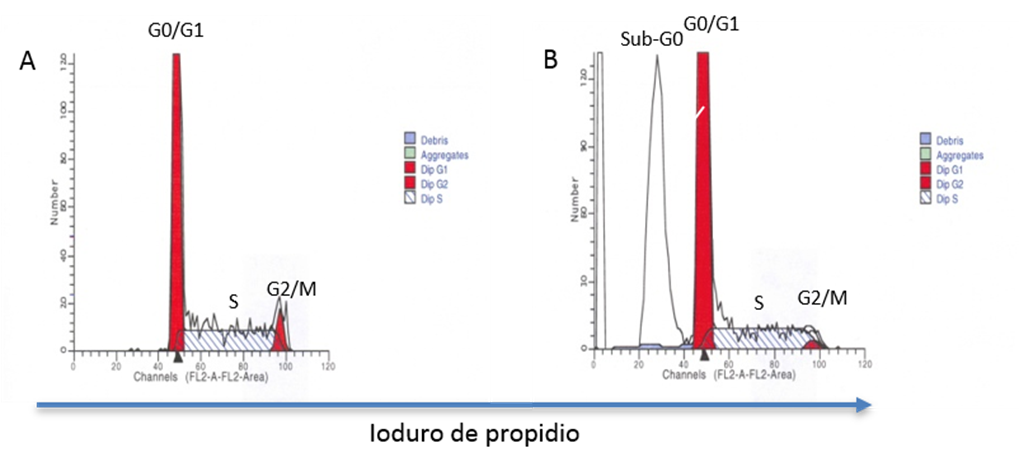
Otra técnica empleada es el análisis de ADN. En este caso se utilizan moléculas que se unen al ADN de manera estequiométrica y que por lo tanto fluorescen con una intensidad que es proporcional al contenido de ácidos nucleicos(64), de tal manera que las células que están en fase G0 o G1 del ciclo celular se ubicarán en un valor en la escala, el cual será la mitad del valor en el cual se ubiquen las células en fase G2 o mitosis (M) temprana, previo a la citocinesis. Las células que están en las diferentes etapas de la fase S, es decir, desde que inicia la duplicación del material genético hasta que termina, se ubicarán entre la posición de las células en G0/G1 y las G2/M (Figura 5A). Cuando una población celular está proliferando, como ocurre en cáncer, se incrementará el porcentaje de células en fase S y en G2/M con respecto a la muestra testigo en la cual no hay proliferación. En este caso, también es necesaria la permeabilización de las células para que el fluorocromo pueda entrar al núcleo y unirse al ADN. Entre las moléculas más utilizadas están el PI, el 7-amino-actinomicina D (7-AAD) y el Hoechst 33342. El estudio del ciclo celular puede extenderse y completarse con el análisis de moléculas involucradas en cada una de las fases del ciclo haciendo uso del potencial multiparamétrico de la CF. Cabe mencionar que con este tipo de análisis también puede identificarse la presencia de células que tienen el ADN fragmentado, las cuales aparecen en lo que se conoce como “pico Sub-G0” (Figura 5B), y que correlaciona con algunos tipos de muerte celular. Con esta técnica también es posible evaluar la estabilidad del genoma si se compara la ploidía del pico G0/G1 entre células tumorales y células sanas(64,65). Aunado a lo anterior y como se mencionará más adelante, esta técnica permite distinguir con base en su contenido de ADN espermatozoides X de espermatozoides Y.
Quizá la técnica más utilizada para evaluar proliferación es la dilución de la carboxifluoresceína (CFSE, carboxyfluorescein diacetate succinimidyl ester). Para esta, se utiliza el compuesto CFDA, el cual no es fluorescente, es lipofílico y puede penetrar la membrana de las células; una vez en el interior, la acción de las esterasas de las células vivas lo convierten en CFSE que es fluorescente, esta molécula se une a las proteínas celulares, dejando marcadas a todas las células de la muestra; si las células proliferan, cada una de las células hijas tendrá la mitad del fluorocromo que tenía la célula de la cual proviene, de tal manera que es factible establecer el porcentaje de células que proliferaron y los ciclos de proliferación con base en la dilución o disminución progresiva de la marca con cada división celular(66).
Figura 5: Análisis del contenido de ADN
Las células se fijan y se tiñen con Ioduro de propidio. El análisis de ADN permite cuantificar el porcentaje de células en las diferentes fases de ciclo celular: G0/G1, S y G2/M con base en el contenido relativo de ADN. La intensidad de la tinción es directamente proporcional a la cantidad de ADN que tienen las células en cada fase (A). Este tipo de análisis permite cuantificar, además, el porcentaje de células que tienen ADN fragmentado, pico identificado como Sub-G0 (B).
Adicional a lo anterior, se puede detectar la expresión de Ki67, una molécula que se ubica en el núcleo y que está asociada a la proliferación celular utilizando un anticuerpo dirigido contra ella como ha sido propuesto en el OMIP-065(14), que es el primer panel de tinción para CF optimizado específico para perros. La detección de Ki67 se ha asociado a los cánceres caninos más agresivos, por lo que puede tomarse como un biomarcador pronóstico(67).
Producción de citocinas. Las citocinas son moléculas solubles que se sintetizan en respuesta a un estímulo y tienen como función principal la comunicación entre las células. Existen dos estrategias generales para detectar la producción de citocinas, una las detecta intracelularmente, lo que permite hacer de manera simultánea la identificación de las células productoras(68) y otra, en la que se pueden cuantificarlas en solución una vez que han sido liberadas en el microambiente en el que se encuentren (plasma, suero u otro líquido corporal, o bien en el medio de cultivo si se trata de un sistema in vitro). En esta última estrategia, no se puede saber qué célula las produjo, a menos que las células hayan sido previamente purificadas. Para el primer caso, a las células debe agregarse un reactivo que impide la secreción de las citocinas como la brefeldina o monensina, posteriormente se permeabilizan y se agregan anticuerpos marcados con fluorocromos dirigidos contra las citocinas que se quieren determinar(69). En el segundo caso, la detección se hace en solución, ya sea en algún líquido corporal o en el medio de cultivo. Para esto, se utilizan partículas que tienen dos características, emiten fluorescencia y tienen anticuerpos dirigidos contra la citocina o molécula de interés pegados en su superficie, los cuales actuarán como anticuerpos de captura (Figura 6A). Las partículas, o perlas como también se les conoce, tienen una intensidad de fluorescencia particular dependiendo de la molécula que reconozcan los anticuerpos sobre su superficie, de tal manera que se pueden detectar varias moléculas solubles en la misma muestra(7).
La cuantificación de las moléculas solubles se realiza con base en la intensidad de fluorescencia emitida por un anticuerpo reportero que se agrega al final y que está acoplado a un fluorocromo diferente al de la perla; en consecuencia, la intensidad de fluorescencia del anticuerpo reportero es directamente proporcional a la concentración de la molécula en estudio (Figura 6B-D). Gracias a la utilización de estándares de concentración conocida de cada molécula que se quiere cuantificar, se puede obtener la concentración presente en la muestra. Con base en lo anterior, el análisis de los resultados es biparamétrico: la intensidad de la perla identifica la molécula que se quiere cuantificar y la intensidad de fluorescencia del anticuerpo reportero, la concentración de esta en la muestra.
Existen en el mercado paquetes comerciales conocidos como “arreglos de perlas” (bead arrays) o “ensayos múltiples basados en perlas” (bead-based multiplex assays) para cuantificar moléculas en solución. A diferencia de los ensayos de ELISA en los que para cuantificar cada molécula se utiliza un volumen dado de muestra, aquí, con el mismo volumen se pueden analizar múltiples moléculas de manera simultánea(6,70). Actualmente y dada su gran versatilidad y potencial en diversas áreas, se han desarrollado equipos destinados únicamente para la lectura de este tipo de ensayos que pueden detectar incluso hasta 500 moléculas solubles con un volumen máximo de 50 μl de muestra, las cuales pueden ser elegidas y solicitadas al fabricante, con base en la necesidad particular del comprador. Si bien la oferta comercial aún es limitada en el ámbito veterinario, Christopher-Hennings et al., la han propuesto como una excelente alternativa para la cuantificación de citocinas, quimiocinas, hormonas, patógenos y anticuerpos en el ámbito veterinario(71). Como ejemplo de ello, se puede mencionar el diagnóstico serológico de Leishmaniasis en perros, en el que las perlas en lugar de anticuerpos de captura, tienen pegados antígenos recombinantes del parásito y lo que se detecta en la muestra son anticuerpos por lo que los anticuerpos reporteros reconocen los anticuerpo de caninos presentes en la muestra(72).
Figura 6: Arreglos de perlas
Los más comunes emplean partículas inertes fluorescentes (círculo rojo) que tienen pegados anticuerpos de captura para la molécula que se desea cuantificar (A). Si la molécula soluble que se quiere cuantificar (círculo verde) está presente en la muestra, se unirá a los anticuerpos. En el último paso, se adiciona un anticuerpo “reportero” que está unido a un fluorocromo (estrella naranja) diferente al de la perla y por lo tanto la intensidad de fluorescencia de este, será proporcional a la cantidad de molécula a cuantificar. En esta aplicación se cuenta con estándares de concentración conocida por lo cual es una técnica cuantitativa. En B, C y D se esquematizan tres posibles resultados de un arreglo de perlas. Muestra que no tiene la molécula a cuantificar (A), muestra que tiene la molécula, pero en poca cantidad (C) y muestra que tiene una alta concentración de la molécula (D). La concentración es directamente proporcional a la intensidad de fluorescencia emitida por el anticuerpo reportero. En un mismo ensayo se utilizan perlas que tienen anticuerpos de captura para diferentes moléculas. Imagen creada con BioRender.
Sexado de espermatozoides. Con base en un análisis de ADN es factible diferenciar en una muestra de esperma de cualquier especie animal, cromosomas X de cromosomas Y, ya que los primeros tienen mayor contenido de material genético. Se estima que, en términos generales, esta diferencia es de 3 a 4 % aproximadamente, y dada la alta sensibilidad que tienen los citómetros de flujo, es factible diferenciarlos e incluso separarlos físicamente en diferentes tubos gracias a la función de “sorter” que tienen algunos equipos y que alcanza hasta más de 90 % de pureza. Los espermatozoides separados, se ha utilizado en inseminación artificial con resultados variados dependiendo de la especie animal(73,74). No hay que olvidar que a los espermatozoides pueden evaluárseles varios de los parámetros antes mencionados como la expresión de moléculas en su superficie o intracelularmente, la integridad de su membrana, la presencia de moléculas involucradas con alguno de los tipos de muerte celular, etc., por lo que los estudios que pueden hacerse a los espermatozoides van más allá que el análisis de ADN, permitiendo hacer estudios sobre la calidad y función de estos(19).
Comentarios finales
El principal uso de la CF en el ámbito veterinario actualmente es en investigación; las aportaciones que se han hecho mediante la utilización de esta tecnología han tenido un gran impacto en el conocimiento de las células, órganos y sistemas en condiciones fisiológicas y patológicas en diferentes especies. El empleo de citómetros de flujo en el área clínica tiene muchas ventajas dada su gran sensibilidad, especificidad y versatilidad en cuanto a pruebas que pueden realizarse; el costo de los equipos y de los reactivos, además de la falta de disponibilidad de muchos de ellos, ha llegado a ser un obstáculo para su implementación en México en medicina veterinaria. Afortunadamente ambas situaciones se han ido superando gracias a la gran demanda que tienen estos equipos en diferentes ámbitos, lo que ha disminuido su precio y se ha ampliado la disponibilidad de reactivos, haciendo poco a poco más accesible su empleo y abriendo la posibilidad de su introducción en los laboratorios clínicos veterinarios de nuestro país. En México, existen diversas alternativas de capacitación en CF tanto teórica como práctica, los cuales son impartidos en algunas universidades del país, así como en el Laboratorio Nacional de Citometría de Flujo (LabNalCit) y en el Capítulo de Citometría de la Sociedad Mexicana de Inmunología, este último pionero en la enseñanza y difusión del uso de los citómetros de flujo y de la correcta interpretación de los resultados obtenidos con esta tecnología.
Conflicto de intereses
Los autores declaran que no existe conflicto de intereses.
Literatura citada: