https://doi.org/10.22319/rmcp.v11i4.5302
Artículo
Análisis genómico de diversidad y estructura genómica de las poblaciones bovinas de la raza mexicana de Lidia
Paulina G. Eusebi a*
Oscar Cortés a
Susana Dunner a
Javier Cañón a
a Universidad Complutense de Madrid. Facultad de Veterinaria, Departamento de Producción Animal. Avenida Puerta de Hierro, s/n, 28040, Madrid, España.
* Autor de correspondencia: paulig01@ucm.es
Resumen:
La raza bovina de Lidia ha sido seleccionada desde el siglo XIII para participar en festejos sociales reconocidos bajo el término de “Tauromaquia”. En la actualidad forman parte de la identidad de las culturas regionales de varios países. En México, la raza se especializó a finales del siglo XIX cuando cuatro familias mexicanas importaron un número reducido de bovinos de España. De estas importaciones actualmente solo permanecen las líneas derivadas de las familias Llaguno y González. En la familia Llaguno se llevaron diferentes estrategias de reproducción. Antonio Llaguno cruzó los recién importados bovinos españoles entre sí; de dichas cruzas derivó la línea que actualmente es reconocida como “Pura”. Por otro lado, Julián Llaguno realizó cruzas entre hembras criollas con machos españoles, línea conocida como “Impura”. Por último, entre1996 y 1997, un grupo de ganaderos importó bovinos pertenecientes a ciertos encastes españoles como Domecq, Murube, Santa Coloma, entre otros. El objetivo del presente estudio fue investigar la diversidad genómica, estructura poblacional, niveles de endogamia y relaciones genéticas entre las poblaciones de Lidia mexicana, utilizando marcadores moleculares de tipo SNP. La población fue dividida en cinco grupos: Antonio Llaguno, Julián Llaguno, González, y dos grupos que incluían bien importaciones recientes de origen Domecq, o bien importaciones recientes de origen Santa Coloma. Los resultados permiten apreciar diferentes orígenes genéticos dentro de la familia Llaguno en función de su origen histórico: Antonio y Julián. En el resto de grupos también se observa una clara diferenciación genética. Este aislamiento genético entre poblaciones de Lidia mexicana es una característica de su singularidad.
Palabras clave: Raza de Lidia, Genética de poblaciones, Diversidad genética, Estructura genética.
Recibido: 23/02/2019
Aceptado: 07/10/2019
Introducción
La raza bovina de Lidia, seleccionada por caracteres de comportamiento que potencian la agresividad, ha sido utilizada desde el siglo XIII en la Península Ibérica para participar en festejos de índole civil y religioso(1). En la actualidad dichas prácticas forman parte de un fenómeno social reconocido como “Tauromaquia”, el cual hace referencia a todas aquellas representaciones culturales y subjetivas que involucran ganado bovino(2,3). Hoy en día varios países consideran a las diferentes tauromaquias como prácticas que refuerzan la identidad de las culturas regionales(2,3); inclusive, en países como España y Perú se le ha nombrado patrimonio cultural inmaterial(4). Además, esta raza se caracteriza por una baja intercambiabilidad genética y ecológica(5,6).
En México el primer festejo taurino documentado ocurrió en 1523, utilizando bovinos de comportamiento agresivo que provenían principalmente de la región de Navarra, donde procede el ganado bovino de Casta Navarra(7), pero no fue, sin embargo, hasta finales del siglo XIX cuando comenzó la cría especializada de la raza en México, al importar un número reducido de animales de la raza de Lidia de España por cuatro familias de criadores: Llaguno, González, Barbabosa y Madrazo(8,9). Actualmente sólo se tiene evidencia de la permanencia de las líneas derivadas de dos de estas familias: Llaguno y González(8,10).
Dentro de la familia Llaguno, localizada en la región centro-norte de la República Mexicana, Antonio Llaguno, mantuvo un sistema cerrado de reproducción cruzando solamente los animales de la raza de Lidia importados entre sí; a los animales derivados de esta línea se les denomina actualmente en el medio ganadero mexicano como “Puros”. Por otro lado su hermano Julián Llaguno llevó a cabo otra estrategia de reproducción cruzando vacas criollas con toros de Lidia de origen español; a los animales derivados de ésta línea hoy se les denomina como “Impuros”(8,9).
Por otro lado, la familia González, ubicada en la región centro-sur de la República Mexicana, cruzó los animales de la raza de Lidia importados con bovinos locales seleccionados por su agresividad(8); por último, entre 1996 y 1997, un grupo de ganaderos mexicanos importó animales pertenecientes a ciertos encastes españoles, entre los que se encuentran Domecq, Murube, Santa Coloma y Saltillo, entre otros(10), posteriormente se cerraron las fronteras a importaciones por cuestiones sanitarias(10).
Actualmente el censo de la población bovina de Lidia mexicana asciende aproximadamente a 110,000 animales distribuidos en un área de alrededor de 135,000 ha(10). Este tipo de ganado es criado en condiciones de producción extensivo, lo cual favorece la conservación de especies endémicas tanto de flora como de fauna. Los festejos en los que se utiliza la raza Lidia en México tienen un papel clave en la economía pecuaria mexicana, formando parte de las tradiciones sociales que refuerzan la identidad de las comunidades locales(7,10,11).
En estudios anteriores se analizó la variabilidad genética de la población Lidia mexicana con respecto a la población original Española por medio de marcadores autosómicos de tipo microsatélite, encontrando una diferenciación entre encastes españoles y las familias mexicanas Llaguno y González(12). Resultados que fueron confirmados cuando la información molecular que se utilizó fue la obtenida mediante chips de ADN para marcadores bi-alélicos tipo SNP. En este caso, además, se observó una clara diferenciación genética entre familias Mexicanas(13,14). Sin embargo, dichos análisis no incluían animales de procedencia Julián Llaguno o denominados “Impuros”, así como tampoco bovinos procedentes de las importaciones recientes de encastes mayoritarios españoles como Domecq y Santa Coloma.
El objetivo del presente estudio fue investigar la diversidad genómica, estructura poblacional, niveles de endogamia y relaciones genéticas entre las poblaciones representativas de la raza de Lidia mexicana, utilizando marcadores moleculares de tipo SNP.
La población fue dividida en función a su origen histórico dentro de cinco grupos: Antonio Llaguno, Julián Llaguno, González, y dos grupos que incluían bien importaciones recientes de origen Domecq e importaciones recientes de origen Santa Coloma.
Material y métodos
Un total de 306 muestras de sangre fueron recolectadas aleatoriamente de bovinos pertenecientes a 32 ganaderías mexicanas afiliadas a la Unión de Criadores de Toros de Lidia, agrupadas históricamente en cinco grupos (Cuadro 1). Las muestras se recolectaron en tubos con conservante Magic Buffer® (Biogen Diagnostica, España) y se mantuvieron a 15° C hasta su uso para la extracción del ADN. El ADN genómico se extrajo mediante un protocolo estándar de fenol/cloroformo(15), posteriormente las muestras se genotiparon con el Chip bovino de SNPs de mediana densidad 50K (http://www.illumina.com).
Cuadro 1: Número de animales analizados (N), diversidad génica (He), heterocigosis observada (Ho), nivel de consanguinidad (FIS) y distancia genética promedio FST de cada ganadería con respecto al resto de ganaderías dentro de grupo, así como de cada ganadería
Origen | Ganadería | N | FST | FIS | HO | HE |
Julian llaguno | Pozo Hondo | 21 | 0.05 | 0.13 | 0.27 | 0.73 |
| Valparaiso | 15 | 0.06 | 0.19 | 0.25 | 0.75 |
| El Sauz | 8 | 0.07 | 0.21 | 0.24 | 0.76 |
| Caparica | 11 | 0.05 | 0.17 | 0.26 | 0.74 |
| Total | 55 | promedio 0.06 |
|
|
|
Antonio Llaguno | San Mateo | 6 | 0.09 | 0.21 | 0.24 | 0.76 |
| Reyes Huerta | 39 | 0.06 | 0.21 | 0.24 | 0.76 |
| Fernando de la Mora | 6 | 0.10 | 0.04 | 0.30 | 0.70 |
| Los Cues | 7 | 0.07 | 0.29 | 0.22 | 0.78 |
| Garfias | 6 | 0.08 | 0.26 | 0.23 | 0.77 |
| Antigua | 6 | 0.09 | 0.27 | 0.23 | 0.77 |
| Xajay | 6 | 0.05 | 0.15 | 0.26 | 0.74 |
| Teófilo Gómez | 6 | 0.07 | 0.19 | 0.25 | 0.75 |
| Celia Barbabosa | 6 | 0.06 | 0.15 | 0.26 | 0.74 |
| Boquilla del Cármen | 6 | 0.06 | 0.29 | 0.22 | 0.78 |
| Fermín Rivera | 6 | 0.07 | 0.18 | 0.25 | 0.75 |
| Corlomé | 6 | 0.13 | 0.00 | 0.31 | 0.69 |
| Arroyo Zarco | 6 | 0.05 | 0.18 | 0.26 | 0.74 |
| Marrón | 6 | 0.05 | 0.11 | 0.28 | 0.72 |
| La Punta | 19 | 0.10 | 0.11 | 0.27 | 0.73 |
| Total | 137 | promedio 0.07 |
|
|
|
González | Tenexac | 8 | 0.13 | 0.29 | 0.22 | 0.78 |
| Yturbe | 5 | 0.11 | 0.14 | 0.27 | 0.73 |
| De Haro | 6 | 0.08 | 0.12 | 0.27 | 0.73 |
| Castañeda | 6 | 0.11 | 0.25 | 0.23 | 0.77 |
| Zacatepec | 12 | 0.12 | 0.03 | 0.30 | 0.70 |
| Rancho Seco | 6 | 0.14 | 0.05 | 0.29 | 0.71 |
| Total | 43 | promedio 0.11 |
|
|
|
Importaciones recientes Domecq | La Joya | 17 | 0.10 | 0.15 | 0.26 | 0.74 |
| Santa Ma. De Xalpa | 17 | 0.08 | 0.06 | 0.29 | 0.71 |
| Jaral de Peñas | 17 | 0.06 | -0.03 | 0.32 | 0.68 |
| Torreon de Cañas | 6 | 0.09 | -0.02 | 0.32 | 0.68 |
| Jose Julian Llaguno | 10 | 0.07 | 0.00 | 0.31 | 0.69 |
| Total | 106 | promedio 0.08 |
|
|
|
Importaciones recientes Santa Coloma | Los Encinos | 5 | 0.02 | 0.17 | 0.26 | 0.74 |
| San José | 6 | 0.02 | 0.10 | 0.28 | 0.72 |
| Total | 11 | promedio 0.02 |
|
|
|
Por medio del Software PLINK V 1.07(16) se depuró la información, excluyendo a los SNP localizados en cromosomas sexuales, aquellos que presentaron una frecuencia del alelo menos común (MAF), inferior a 0.01, con menos del 20 % de genotipos faltantes y aquellos marcadores que se separaban del equilibrio de Hardy-Weinberg (P<0.001). Finalmente se obtuvieron 41.455 SNP para su análisis.
Se analizaron parámetros de diversidad genética tales como heterocigosis observada (Ho), heterocigosis esperada (He) y coeficiente de endogamia (FIS) estimado como1-Ho/He mediante el software PLINK V 1.07(16). Los coeficientes de FST se calcularon por medio del software ARLEQUIN v3.0(17). Para cada individuo se obtuvo la proporción de orígenes genéticos que se podían identificar utilizando el algoritmo de agrupamiento bayesiano implementado en el software ADMIXTURE(18,19). Las gráficas se realizaron con el Software POPHELPER v1.0.10(20).
Se llevó a cabo también un Análisis Molecular de Varianza (AMOVA) mediante un modelo lineal(17) para evaluar la variación genética entre y dentro de grupos. Se realizó el análisis en un modo jerárquico con tres niveles (entre grupos, entre ganaderías dentro de grupos y dentro de ganaderías). También se calculó la distancia promedio de los grupos en términos de FST utilizando el mismo software.
Finalmente, se identificaron los ROHs (Runs of Homozygosity) individuales por individuo(21), utilizando Plink v.1.07 con ventanas de 30 SNP, permitiendo <100 kb entre dos SNP homocigotos consecutivos, menos de dos genotipos faltantes, un heterocigoto, y con una longitud mínima de 500 kbp. Posteriormente se obtuvo el valor promedio por ganadería y por grupo.
Resultados y discusión
Diversidad genética
En el Cuadro 1 se presentan los resultados de diversidad génica (He), heterocigosis observada (Ho) así como valores de consanguinidad (FIS) y la distancia genética FST de cada ganadería con respecto al resto de ganaderías dentro de cada grupo. Se puede observar que los valores promedio de FIS por ganadería oscilaron entre -0,03 (Jaral de Peñas) y 0.29 (Los Cues, Boquilla del Cármen y Tenexac). El exceso de heterocigotos de las ganaderías de Jaral de Peñas y Torreón de C., pertenecientes al grupo de “Importaciones recientes origen Domecq”, explica los valores negativos de FIS como consecuencia del efecto Wallhund(22). Las distancias promedio FST estimadas por grupo fueron similares entre ellos, con valores de entre 0.06 y 0.11, mientras que los valores de las distancias FST promedio estimadas por ganadería oscilaron entre 0.02 (Los Encinos y San José) y 0.14 (Rancho Seco). En la población mexicana existe la práctica entre ganaderos de intercambiar hembras y sementales, siendo una práctica más común entre ganaderos pertenecientes a los mismos grupos u orígenes. Si bien, la frecuencia y cantidad de animales en dichos intercambios depende del criterio del ganadero, esto va a explicar que las distancias genéticas no difieran mucho entre ganaderías dentro del mismo grupo.
El AMOVA evidenció que el 10.8 % de la variabilidad genética total se debe a las diferencias entre grupos, que las diferencias entre las ganaderías dentro de los grupos explican un 6,9% (Cuadro 2). Es de esperar que al no intercambiar individuos entre grupos la diferencia entre estos sea mayor que dentro de grupos, donde se dan intercambios de forma más regular. El valor FST promedio entre grupos (0.18) es similar al valor FST promedio obtenido en la población española de Lidia (0.15) y mayor al observado en otras razas bovinas con valores alrededor de 0.07(6). Los valores elevados de FST son consecuencia de la estructura que caracteriza a la raza de Lidia, en la que la subdivisión en subpoblaciones o grupos resulta en tamaños efectivos pequeños por grupo.
Cuadro 2: Resultados del análisis molecular de varianza ANOVA
Nivel | Componente de varianza | % de variación |
Entre grupos | 686.54 | 10.76 |
Entre ganaderías dentro de grupos | 437.74 | 6.86 |
Dentro de ganaderías | 5259.13 | 82.39 |
Residual | 6383.41 |
Estructura genética y diferenciación de la población
El error de validación cruzada (CV) utilizado en el Software ADMIXTURE determina valores que disminuyen al aumentar el número de poblaciones ancestrales hipotéticas (K). En el momento en que dicho valor de CV comienza a aumentar es indicativo de la predicción de poblaciones hipotéticas más probable. En este caso la predicción más acertada se identificó en k=5(18,19).
Las proporciones promedio de los individuos de cada ganadería asignada a cada una de las 5 poblaciones ancestrales se presentan en la Figura 1 en donde se agruparon de acuerdo al origen las ganaderías. Cada uno de los cinco grupos se asocia mayoritariamente a cada una de las cinco poblaciones ancestrales definidas. No obstante, entre el grupo “Julián Llaguno” y “Antonio Llaguno” la discriminación es menos evidente entre algunas ganaderías de ambos grupos, mientras que, en otras, como El Sauz y Valparaíso (Julián Llaguno) o Garfias, Los Cués y La Antigua (Antonio Llaguno) la diferenciación es mayor. Aunque el análisis realizado no permite conocer con precisión las causas de la ligera diferenciación entre algunas ganaderías en el caso de la Familia Llaguno (Antonio y Julián) podría ocurrir que diferencia entre ambos grupos se deban al diferente material genético en origen, siendo Julián quien cruzó hembras criollas con machos de Lidia españoles. En todo caso, se puede observar que ambos grupos (Antonio y Julián Llaguno) comparten mayoritariamente orígenes genéticos comunes. Por otro lado, en los grupos de Gonzáles y las importaciones recientes origen Domecq y Santa Coloma sí que se puede observar una clara diferenciación genética entre grupos.
Figure 1: Análisis de errores de validación cruzada de poblaciones ancestrales hipotéticas (K) utilizando ADMIXTURE
Cada línea vertical representa el genoma total por individuo, y la proporción que de cada color (grupo genético, K) hay en dicha línea vertical, es la proporción del genoma de ese individuo representado de acuerdo a los cinco orígenes ancestrales (K).
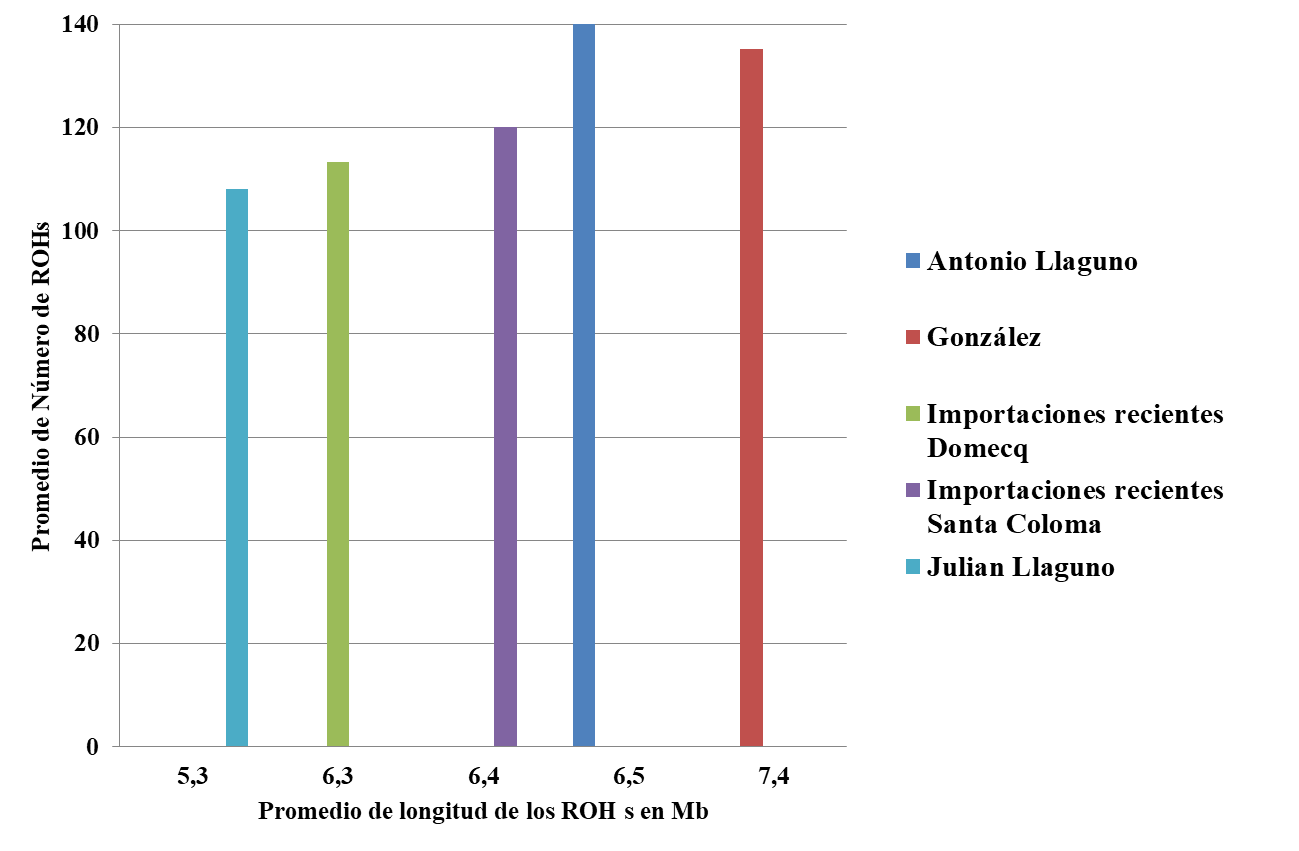
Finalmente se procedió a identificar las regiones de homocigosis o ROHs por grupo. En la Figura 2 se muestra la estadística sobre el número promedio de regiones o segmentos y la longitud promedio de los ROH en cada uno de los cinco grupos. En general se observan patrones similares en cuánto a número y longitud de los ROH entre los diferentes grupos. Se sabe que un mayor número de segmentos y longitud de los ROH están correlacionados con eventos de consanguinidad reciente(23).
Figura 2: Número promedio de regiones de homocigosis (ROH) y tamaño promedio en Mb de los cinco grupos de la población mexicana de Lidia
En el Cuadro 3 figura el número y tamaño de ROHs promedio por ganadería, observando que la diferencia entre la longitud promedio de ROHs oscila entre 5 (Jaral de Peñas) y 7.7 Mb (Boquilla del Carmen y el Sauz), lo cual es coherente con los valores promedios de FIS obtenidos (Cuadro 1) dónde los valores más altos (>0.20) corresponden a las ganaderías de Boquilla del Carmen y el Sauz y los más bajos a la ganadería de Jaral de Peñas.
Cuadro 3: Regiones de homocigosis (ROH) promedio por ganadería; número de segmentos (NSEG) y longitud promedio en Mb
Origen | Ganadería | NSEG | Mb. promedio |
Julián Llaguno | Pozo Hondo | 108 | 5.3 |
| Valparaiso | 134 | 6.2 |
| El Sauz | 111 | 7.7 |
| Caparica | 114 | 6.5 |
| Pomedio | 117 | 6.1 |
Antonio Llaguno | San Mateo | 141 | 6.5 |
| Reyes Huerta | 124 | 5.9 |
| Fernando de la Mora | 87 | 5.8 |
| Garfias | 139 | 7.1 |
| Los Cués | 131 | 7.3 |
| La Antigua | 144 | 6.9 |
| Xajay | 115 | 6.6 |
| Teófilo Gómez | 135 | 6.0 |
| Celia Barbabosa | 126 | 5.8 |
| Boquilla del Cármen | 132 | 7.7 |
| Fermín Rivera | 131 | 6.1 |
| Corlomé | 79 | 5.3 |
| Arroyo Zarco | 124 | 6.3 |
| Marrón | 105 | 6.2 |
| La Punta | 104 | 6.1 |
| Pomedio | 121 | 6.2 |
González | Tenexac | 135 | 7.4 |
| Gonzalo Yturbe | 110 | 6.3 |
| De Haro | 113 | 5.6 |
| C.Castañeda | 134 | 6.9 |
| Zacatepec | 83 | 5.7 |
| Rancho Seco | 95 | 5.2 |
| Pomedio | 109 | 6.2 |
Importaciones recientes Domecq | La Joya | 113 | 6.3 |
Sta. Maria de Xalpa | 92 | 5.7 | |
| Jaral de Peñas | 64 | 5.0 |
| José Julián Llaguno | 72 | 5.1 |
| Torréon de Cañas | 73 | 5.2 |
| Pomedio | 85 | 5.5 |
Importaciones recientes Santa Coloma | Los Encinos | 120 | 6.4 |
San José | 105 | 5.8 | |
| Pomedio | 112 | 6.1 |
En situaciones de aislamiento, en poblaciones con tamaño relativamente pequeño, se incrementa la probabilidad de que los animales hereden segmentos idénticos de ADN que justifican la presencia de ROH(23). En estudios previos se han asociado el elevado número y longitud de los ROH a prácticas endogámicas(23), lo cual concuerda con los resultados obtenidos para los cinco grupos mexicanos de Lidia, en los que se observan valores mayores a los observados en análisis previos en los que se analizan regiones de homocigosis en razas autóctonas españolas y criollas americanas(13).Tanto los valores de los ROHs como de FIS en la población de Lidia mexicana son reflejo de la subdivisión en grupos, y sus consecuencias: reducción de tamaños efectivos e incremento en los valores de consanguinidad(24). Aunque esta subdivisión es una alternativa en la preservación de la variabilidad genética de una población(25), en cada subpoblación, como consecuencia de la deriva genética se produce un aumento de la consanguinidad y pérdida de la variabilidad genética, lo que aconseja monitorizar la evolución de la endogamia.
Conclusiones e implicaciones
La diferenciación genética de la población mexicana entre grupos e inclusive entre ganaderías se debe o bien a los diferentes orígenes genéticos de algunos grupos (importaciones recientes Domecq y Santa Coloma), o debido al aislamiento genético que se ha mantenido en los grupos restantes (Antonio y Julián Llalguno y el grupo González). Tanto los análisis de estructura genética como el comportamiento de los ROH revelan que el aislamiento genético de los grupos que componen a la población mexicana de Lidia ha contribuido a las variaciones genómicas con respecto a poblaciones de ganado de Lidia europeo; reflejando su grado de singularidad.
Agradecimientos
La financiación de este trabajo proviene del “Macro-proyecto de Caracterización genética de la Raza de Lidia Mexicana”, realizado con apoyo del Consejo Nacional de los Recursos Genéticos Pecuarios (CONARGEN) y la Asociación de Criadores de Toros de Lidia. También se agradece al Laboratorio de Genética del Departamento de Producción Animal de la Facultad de Veterinaria en la Universidad Complutense de Madrid, y a los ganaderos mexicanos de Lidia.
Literatura citada: