https://doi.org/10.22319/rmcp.v11i4.5295
Nota de investigación
Análisis de QTL asociados a polimorfismos de nucleótido único (SNP) involucrados en el fenotipo lechero del ganado Holstein
Jose Manuel Valdez-Torres a
Juan Alberto Grado Ahuir a
Beatriz Elena Castro-Valenzuela a
M. Eduviges Burrola-Barraza a*
a Universidad Autónoma de Chihuahua. Facultad de Zootecnia y Ecología. Periférico Francisco R. Almada Km 1, C.P. 31453. Cd. Chihuahua, Chih. México.
*Autor de correspondencia: mburrola1@uach.mx
Resumen:
El objetivo fue identificar QTL asociados a polimorfismos de nucleótido único (SNP) cuya acción contribuya al desarrollo fenotípico productivo, reproductivo y de salud del ganado lechero de raza Holstein. Ubicados en 120 genes del genoma Bos taurus UMD_3.1.1, se identificaron 341 QTL asociados a 189 SNP con efectos sobre rasgos productivos (FY, NM, MY, MTCAS, MBLF y PL), reproductivos (CONCRATE, DPR, EMBSUR, DAYOPEN y CONCEPT) y de salud (SCC, BTBS y RESRATE). Los SNP fueron verificados en la base de datos dbSNP-NCBI donde 42 % se ubicaron en intrones. Con la plataforma Jvenn se supo que los SNP rs135744058, rs110828053 y rs109503725, fueron comunes en los tres rasgos. La red de correlaciones entre rasgos y genes generada por MetScape (Cytoscape 3.4), mostró una correlación positiva entre PL, DPR, DAYOPEN, CONCRATE y CONCEPT; y una negativa de FY hacia PL, NM, DPR y CONCRATE. La funcionalidad de cada gen fue validada en las bases Gene-NCBI y UniProt, y por ClueGo (Cytoscape 3.4) se seleccionaron vías funcionales con un valor de significancia menor a 0.05, lo que evidenció un entrelazamiento entre el desarrollo de la glándula mamaria, la activación del sistema inmune y la respuesta a hormonas esteroideas; siendo el gen GH quien dirige dicha funcionalidad. Aunque el panorama genético muestra que existe un antagonismo entre rasgos productivos y reproductivos, la actividad genética funcional debido a los 189 SNP analizados, muestra una acción entrelazada en vías ontológicas que influyen tanto en los procesos de producción, así como en vías de reproductivas y de salud.
Palabras clave: QTL, SNP, Holstein.
Recibido: 19/03/2019
Aceptado: 17/09/2019
En el ganado Holstein, varios estudios han identificado locus de un carácter cuantitativo (QTL) asociados a rasgos productivos, reproductivos y de salud(1-3). Gracias al perfeccionamiento de los métodos estadísticos y al desarrollo de las herramientas moleculares ha sido posible realizar estudios de asociación del genoma completo (GWAS)(4,5), con lo que se han identificado QTL asociados a un locus, cuya influencia sobre el fenotipo puede variar entre individuos de una misma especie en el cambio de una simple base en el genoma, esto es lo que se conoce como polimorfismo de nucleótido único (SNP). Este tipo de QTL que se correlacionan con un solo SNP tienen un efecto sobre la funcionalidad de un gen específico y por ende una acción inmediata en el desarrollo de un rasgo fenotípico de interés(6). Así que identificar las vías biológicas y los genes que están asociados a SNP significativos, podría dar un conocimiento biológico más profundo sobre el mecanismo de expresión de un determinado rasgo fenotípico(7).
El objetivo de este estudio se centró en buscar con herramientas bioinformáticas, aquellos QTL asociados a un SNP con un potencial efecto sobre los rasgos fenotípicos de producción, reproducción y salud del ganado lechero Holstein. Para la búsqueda de QTL se utilizaron 15 caracteres fenotípicos: duración de la vida productiva (PL), merito neto (NM), rendimiento de leche (MY), rendimiento de proteína de leche (PY), rendimiento de grasa en leche (FY), contenido de caseína (MTCAS), contenido de β-lactoglobulina (MBLG), susceptibilidad a tuberculosis bovina (BTBS), frecuencia respiratoria (RESRATE), contento de células somáticas (SCC), tasa de concepción (CONCRATE), tasa de preñez de las hijas (DPR), sobrevivencia embrionaria temprana (EMBSUR), intervalo parto-concepción (DAYOPEN) y servicios por concepción (CONCEPT). La selección se realizó dentro de los 114,685 QTL reportados en la base de datos en QTLdb (en inglés Animal QTL Database)(2), aquellos QTL asociados a uno de los rasgos productivos y de salud, cuyo pico estuviera relacionado a un solo SNP con un valor de significancia menor a 0.05. Para el caso de los SNP reproductivos, se seleccionaron aquellos reportados por Cochran et al(8) que no tuvieron un efecto negativo sobre algún rasgo productivo. Tomando como base el genoma de Bos taurus UMD_3.1.1, cada SNP se verificó en la base de datos dbSNP del NCBI (en inglés National Center for Biotecnology Inormation, www.ncbi.nlm.nih.gov/snp/), donde se comprobó su ubicación cromosomal, así como el gen afectado y se clasificaron de acuerdo a su localización como: intrón (si estaba en un región intrónica), cambio de sentido (si provocaba un cambio en la secuencia de aminoácidos), sinónimo (si el cambio de base no implicaba un cambio en la secuencia de aminoácidos), promotor (si estaba localizado en el promotor del gen afectado), deleción/inserción (si el SNP resultaba en una deleción o una inserción de una base), UTR3´ y UTR5´ (si el SNP se localizaba en la región no traducida 5´o 3´ del RNAm).
La función de cada uno de los genes implicados en este estudio fue validada tanto en la base de datos Gene del NCBI (www.ncbi.nlm.nih.gov/gene/) como en UniProt (del inglés The Universal Protein Resource, www.uniprot.org). Los SNP comunes entre los rasgos fenotípicos se identificaron a través de un diagrama de Venn utilizando la plataforma Jvenn(9). Mediante el algoritmo de Metscape(10) se calcularon los coeficientes de correlación de Pearson, para establecer asociaciones entre los rasgos fenotípicos con la presencia de un gen. Estos valores fueron visualizados como una matriz colorimétrica (heat map) compuesta de un espectro de color que fue del verde hacia el rojo con valores de correlación del -1 a 1, respectivamente. Estos datos también fueron analizados como un agrupamiento jerárquico y con ellos se generó una de red de correlaciones utilizando la aplicación MetScape del programa Cytoscape 3.4(11). La red funcional ontológica de los genes se realizó con la aplicación ClueGo del programa de Cytoscape 3.4, bajo los siguientes criterios: los datos ontológicos de cada gen se tomaron de la base de datos “GO Biological Process-GOA; con un intervalo “GO Tree” de 3 a 8; los términos de selección para cada vía incluyeron al menos 1 gen por “cluster” con un “kappa score” de 0.3; y se probó con una prueba estadística hipergeométrica bilateral con la corrección de Bonferroni, se tomaron en cuenta sólo aquellas vías con un valor de significancia menor a 0.05.
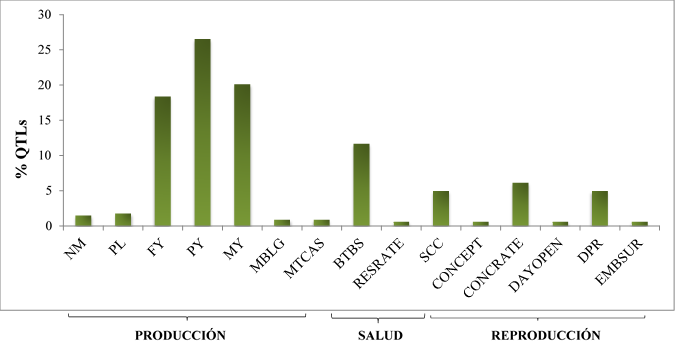
Los resultados publicados a la fecha sobre GWAS en el bovino lechero, han proporcionado información sobre la influencia de los SNP en la expresión de un QTL(4,5,12). En el genoma, los SNP son las formas más abundantes del ADN y debido a su baja tasa de mutación son excelentes marcadores de selección(13,14), además de que es fácil y de bajo costo realizar su genotipificación(15). Tomando la información incluida en la base de datos QTLdb(2), se encontraron 341 QTL asociados a un SNP, de estos el 70 % fueron QTL de producción, 17 % involucrados en la salud y 13 % en rasgos reproductivos (Figura 1). Dentro del fenotipo de producción, el carácter más favorecido fue PY, seguido por MY, luego FY, NM, PL, MBLG y por último MTCAS. En los caracteres de salud, fue BTBS seguido de SCC y RESRATE. En los rasgos reproductivos se presentaron más QTL asociados a CONCRATE, luego a DPR, CONCEPT y el menos favorecido fue EMBSUR. Hubo QTL que tenían el mismo SNP, así como SNP que estaban presentes en diferentes regiones de un mismo gen. Por lo que al final se identificaron 341 QTL asociados a 189 SNP ubicados en 120 genes del genoma Bos taurus. Cada uno de los 189 SNP fueron verificados en la base de datos dbSNP del NCBI, donde se identificó su ubicación y el gen al que afectaban.
Figura 1: Distribución de 341 QTL asociados a un solo SNP, de acuerdo al fenotipo que favorecen
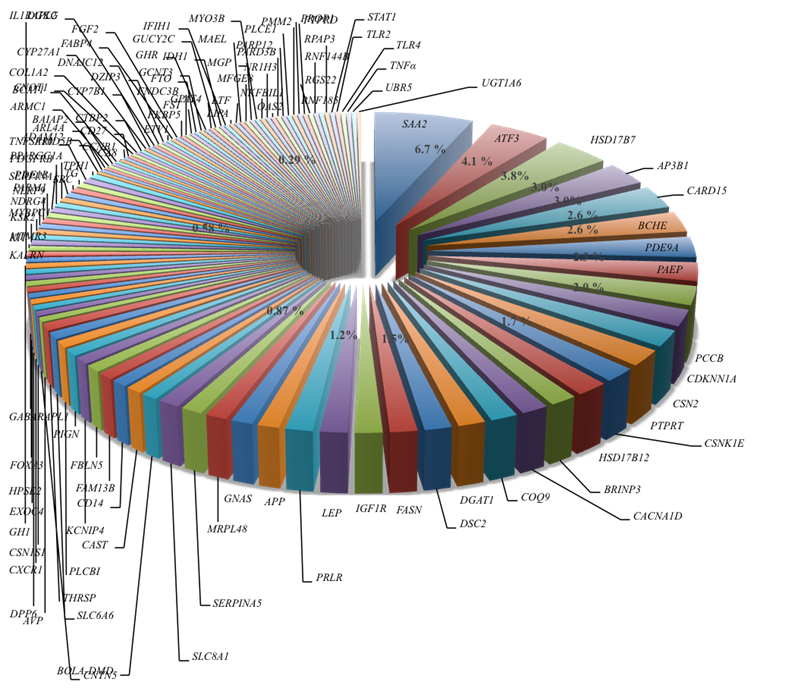
De acuerdo con la información recabada, el 42 % de los SNP fueron intrónicos, 27 % SNP estuvieron ubicados en regiones codificantes, de estos 17 % tuvo efecto en el cambio de aminoácido de la proteína y 5 % fueron mutaciones sinónimas, 6 % se localizaron en el extremo 3´ no traducido (UTR3´) y el 1 % en el UTR5´. El 6.7 % de los SNP estuvieron ubicados dentro del gen SAA2, 4.1 % se localizaron en el gen ATF3, 3.8 % en el gen HSD17B7, 3.0 % estuvieron en cada uno de los genes AP3B1 y CARD15. Los genes BCHE y PDE9A tuvieron el 2.6 %, mientras que dentro de los genes PCC8, CDKN1A y CSN2 se ubicaron en cada uno de ellos un 2 %. En las regiones de los genes APP, GNAS, NRPL48, SERPINA5, SLC8A1, CACNA1D, COQ9, DGAT1, DSC2, FASN, IGF1R, LEP, PRLR, CSNK1E, BRINP3 y HSD17B13 se ubicaron desde un 1.2 al 1.8 % de los SNP (Figura 2). Con excepción de los cromosomas Bta-9 y Bta-12, los 189 SNP se distribuyeron en los todos los autosomas y en el cromosoma X del genoma Bos taurus UMD 3.1.1 (Figura 3). Sin embargo, la distribución fue desigual; hubo cromosomas que presentaron más SNP que otros, y de la misma forma hubo cromosomas que incluyeron pocos SNP, pero estos estuvieron asociados a muchos QTL.
Figura 2: Distribución porcentual de los genes con un SNP asociado a un QTL
Figura 3: Distribución de los rasgos productivos, reproductivos y de salud en el genoma Bos taurus
PY, FY, MY, NM, PL, MBLG, MTCAS, BTBS, SCC, RESRATE, CONCEPT, CONCRATE, DAYOPEN, DPR, EMBSUR
Los QTL que más se identificaron fueron principalmente productivos, lo que apoya el hecho de que la cantidad y calidad de leche producida van de la mano con las ganancias económicas(16). En el Bta-14, el SNP rs109421300 ubicado en el gen DGAT1, tiene más influencia sobre los rasgos MY, FY y PY(14, 15, 17-19). El Bta-5 se asocia a MY, FY y PY con los SNP rs41591907, rs41256890, rs41592943, rs41592948, rs137408198 y rs133449166(7,17,20), ubicados en los genes BCATI, MGP, GUCY2C, GABARAPL1, CD27 y CSNK1E. Bta-18 ha sido asociado tanto a MY como a PY; debido a la influencia del SNP rs41581694(14,17), el cual está ubicado en el gen FOXA3. En el Bta-20, el SNP rs385640152 provoca un cambio en la proteína GHR que afecta MY y PY(7). En el Bta-21, el SNP rs41644615 ubicado en el gen SERPINE5 se asocia con MY y PY(19). Por último se asocia a PY el SNP rs110475419(14), el cual está en el gen ADAM12 en Bta-26.
La leche está constituida por las proteínas α-lactoalbúmina, β-lactoglobulina y las caseínas α, β y κ(21). Se ha asociado el Bta-11 con las concentraciones en la leche de β-lactoglobulina (MBLG), debido al efecto del gen PAEP, que tiene los SNP rs41255679(16,21,22), rs110066229(21) y el rs110180463(21). Por otro lado, Bta-6 ha sido relacionado con MTCAS a través del SNP rs109299401(23) que al expresarse en el gen CSN2, ocasiona un cambio funcional en la β-caseína. MTCAS también se ha asociado a Bta-14 y Bta-19 debido al SNP rs110757796 y rs41923484, respectivamente(23). En el ganado lechero, la enfermedad más común y que más pérdidas económicas acarrea es la mastitis, en donde el conteo de células somáticas (SCC) en leche es un predictor asociado a esta enfermedad(12). Los cromosomas que más influencia tienen en SCC son los Bta-6, Bta-13, Bta-14, Bta-19 y Bta-20(24). En Bta-6 hay 6 SNP, de los cuales rs43703013, rs43703011, rs109299401 están en la región codificante del gen CSN2, donde cada uno de ellos ocasiona un cambio de aminoácido en la proteína β-caseína, mientras que rs110239379 y rs110118210 están ubicados en el mismo intrón del gen KIT; y el rs109757609 se localiza en el promotor del gen CSNIS1. La asociación con SCC en Bta-13 se relacionó con dos SNP en el gen SRC, el rs41703851 y el rs41602996. Bta-14 contribuyó a SCC debido a los SNP rs109162116 y rs109234250 en el gen DGAT1. Otros SNP asociados a SCC son rs109149276 y rs109149276, del gen FASN del Bta-19 y del promotor del gen PRLR en Bta-20, respectivamente(24); y de igual manera, el rs43315150 ubicado en el Bta-2 en un intrón del gen CYP27A1(20).
La tuberculosis es otra enfermedad que provoca pérdidas económicas en la industria pecuaria, además de que siendo el bovino uno de los animales que más convive con el humano, se ha convertido en una de las principales fuentes de diseminación de esta enfermedad a nivel mundial(25). Richardson et al (1) encontraron asociado a BTBS a el Bta-1 con 9 SNP (rs42294486, rs29020933, rs29020933, rs42294431, rs42294441, rs110098599, rs132953892, rs41665131 y rs43741780), localizados en la región promotora del gen BCHE; también 2 SNP (rs109186526 y rs110679397) ubicados en el mismo intrón del gen KALRN. En el mismo trabajo, encontraron que los cromosomas Bta-3, Bta-8, Bta-10 y Bta-23, los SNP rs110622046, rs135916795, rs109277058 y rs41642913 ubicados en regiones intrónicas de los genes MAEL, PTPRD, AP3B1 y FKBP5, respectivamente. Por otro lado Finlay et al(26) reportaron que en el Bta-22 los SNP rs42286978, rs42287005 y rs42724727, que se ubican en la región promotora del gen SLC6A6, se asocian también con la susceptibilidad a tuberculosis. Wang et a (27) reportaron que en el Bta-18, el gen CARD15 presentaba 3 SNP dentro de la región codificante del exón 4 y un SNP localizado en intrón previo al exón 4.
En cuanto a los rasgos reproductivos, estos estuvieron localizados muy cerca de rasgos productivos en los cromosomas Bta-1, Bta-3, Bta-5, Bta-10, Bta-15, Bta-16, Bta-18, Bta-22, Bta-23 y Bta-24 (Figura 3). Algo importante de mencionar es que aunque estos SNP representan sólo una pequeña porción de los genes involucrados en el proceso reproductivo, fueron seleccionados a partir de los trabajos de Cochran et al(8) y Ortega et al(28), quienes demostraron que estos SNP tienen un efecto positivo en rasgos reproductivos asociados con fertilidad, y al mismo tiempo no influyen de manera negativa en los rasgos productivos.
Del total de SNP, solo rs135744058, rs110828053 y rs109503725, fueron comunes entre los rasgos de producción, salud y reproducción. El SNP rs135744058 está asociado con los caracteres MY(8), CONCRATE(8,28), DPR(8) y RESRATE(29). En el idioma genético está compuesto por las variantes A/G(8) y está ubicado en la región exónica del gen CACNAID donde ocasiona el cambio en la subunidad α1D del canal de voltaje de calcio(30). En el bovino se ha detectado su presencia a nivel del hipotálamo durante el desarrollo del sistema nervioso central(31). El SNP rs110828053 ha sido reportado asociado con NM(8), DPR(28), CONCRATE(28), CONCEPT(28) y RESTATE(29); y genera una permuta de A/G en el gen HSD17B7 que provoca un cambio treonina por alanina en la posición 308 de la proteína. El gen HSD17B7 codifica para la deshidrogenasa 7 hidroesteroide 17-β que participa en la biosíntesis de esteroides sexuales y colesterol(28), ha sido localizado en el bovino en el células epiteliales del oviducto(32) siendo esencial para la función ovárica y la regulación de la fertilidad(33). El SNP rs109503725 compuesto por las variables T/C en el gen DSC2, está asociado a los caracteres PL(8), DPR(8,28), CONCRATE(28) y SCC(8). Este SNP genera un cambio del aminoácido 535 de triptófano por arginina en la proteína Desmocolina 2, la cual es una proteína involucrada en las uniones célula-célula conformando los desmosomas en las células epiteliales(34).
Durante las últimas dos décadas en el ganado lechero, las estrategias de selección se han enfocado en desarrollar animales altamente productores de leche, lo que ha traído como consecuencia un declive en la capacidad reproductiva. Esto ha generado que exista una asociación genética negativa, es decir antagonista, entre los rasgos productivos y reproductivos(20,35-37). Ahora bien, en este estudio los SNP reproductivos fueron previamente reportados como no antagonistas con rasgos productivos(8,28); de hecho estos SNP se localizan en las mismas regiones que SNP asociados a rasgos productivos (Figura 3). Con el fin de ubicar aquellos rasgos que se relacionan de manera positiva, se realizó un análisis de correlación de los caracteres fenotípicos en base a los genes que compartían (Figura 4A). Este análisis logró clasificar los caracteres en dos grupos, agrupados en dos clados. El clado 1 agrupó todos los rasgos reproductivos juntamente con los caracteres PL y NM de los rasgos productivos. El resto de los caracteres productivos (MY, PY, FY, MTCAS y MBLG) quedaron agrupados junto con los rasgos de salud en el clado 2 (Figura 4B).
Figura 4: Correlación de los rasgos fenotípicos en base a un SNP asociado a un QTL
(A) Matriz de correlación de Pearson (Heat map), el espectro de color va del verde hacia el rojo con valores de -1 a 1, respectivamente. (B) Agrupación jerárquica de A. DPR= tasas de preñez de las hijas; CONCRATE= tasa de concepción; PL= duración de la vida reproductiva; NM= mérito Neto; DAYOPEN= intervalo parto-concepción; CONCEPT= inseminaciones por concepción; RESRATE= frecuencia respiratoria; EMSUR= sobrevivencia embrionaria temprana; MY= rendimiento de leche; PY= rendimiento de proteína en leche; FY= rendimiento de grasa en leche; SCC= conteo de células somáticas en leche; MTCAS= contenido de caseína en leche; MBLG= contenido de β-lactoglobuina en leche; BTBS= susceptibilidad a tuberculosis bovina.
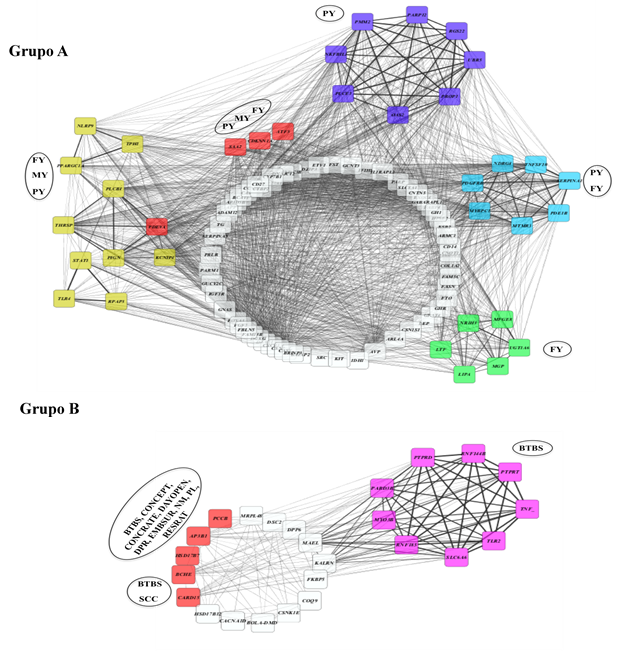
Al analizar la red de correlaciones de los genes en base al rasgo, se pudo observar de la misma manera dos grupos de redes genéticas (Figura 5), el Grupo A correspondiente a aquellos genes presentes en los caracteres FY, MY y PY (rasgos de producción) y el Grupo B con genes presentes en BTBS, SCC, CONCEPT, CONCRATE, DAYOPEN, DPR, EMBSUR, NM, PL, RESRATE abarcando los fenotipos de salud, reproducción y producción. Esto resultados indican que a nivel genético existe un panorama antagonista entre los QTL de producción y reproducción.
Figura 5: Red de correlación de Pearson de los genes asociados a un rasgo fenotípico
Los nodos representan el gen y las líneas negras representan las correlaciones entre los nodos. El análisis se realizó con la aplicación Metscape del programa Cytoscape considerando las positivas de 0.5 a +1. Entre más gruesa sea la línea más alta la correlación. Los nodos en color amarillo, morado, azul, verde y rosa; representan nodos con alta correlación generando grupos y los nodos en color rojo representan a los genes con más SNP implicados en el panel. Las leyendas dentro de los óvalos negros, representan el carácter fenotípico asociado a los genes en cada grupo de nodos.
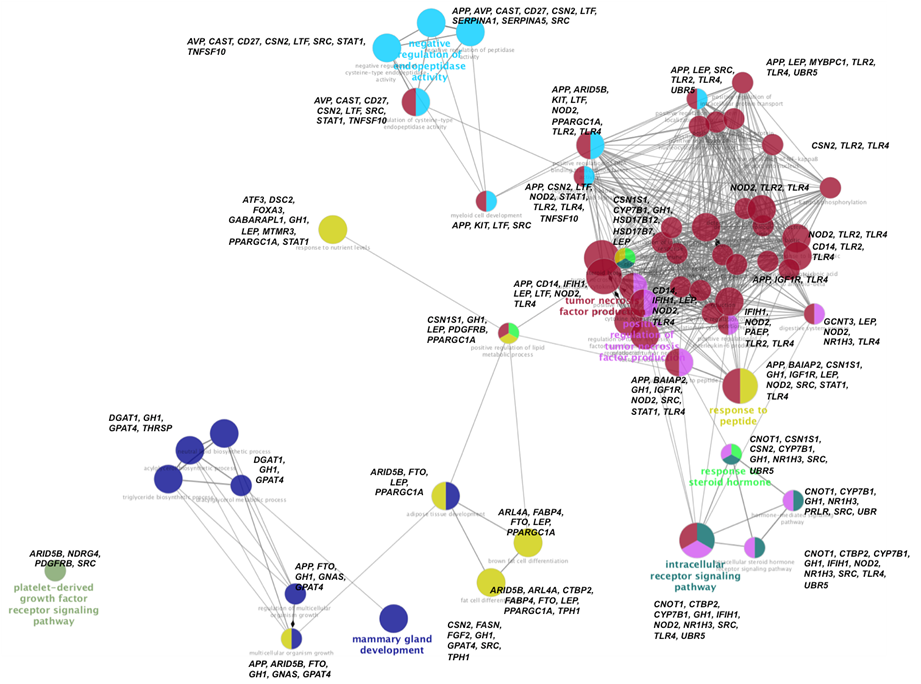
Con el fin de englobar la información funcional de los genes implicados en este estudio, se realizó una red ontológica donde se ubicaron aquellos genes cuyas funciones estuvieran relacionadas entre sí. En la Figura 6 se observa que partiendo del desarrollo de la glándula mamaria donde participan los genes CSN2, FASN, GH, GPAT4, SRC y TPH1; el gen GH enlaza hacia las vías que corresponden al desarrollo de lípidos, donde se encuentran los genes DGAT1, GPAT4 y THRSP. De aquí GH y GPAT4 enlazan hacia vías de crecimiento celular como lo es el desarrollo del tejido adiposo, donde participan los genes APP, ARID5B, FTO, GNAS, GPAT4, LEP, PPARGC1A; de los cuales ARID5B es quien dirige hacia la diferenciación de células grasas, mientras que FTO, LEP y PPARBC1A participan en la diferenciación hacia células grasas marrón. Aquí de nuevo GH, LEP y PPARGC1A son el engranaje para enlazarse junto con CSN1S1 hacia las vías funciones de respuesta a las hormonas esteroides y hacia la regulación del sistema inmune.
Figura 6: Ontología genética (GO) de los genes que componen el Panel representados en una red de funciones biológicas, generada con la aplicación ClueGo del programa Cytoscape 3.4
Cada GO es representado como un nodo circular, cuyo tamaño se relaciona con la significancia (P<0.05) y el número de genes asociados a ese proceso biológico, agrupados por color que involucra un mismo proceso biológico. Los genes implicados en cada GO se encuentran en letras negras.
En la respuesta a las hormonas esteroideas los genes que participan son CSN1S1, CYPP7B1, GH, HSD17B12, HSD17B7 y LEP; de aquí de nuevo GH y CSN1S1 dirigen hacia las vías de señalización de hormonas donde participan CNOT1, NRH13, SRC, UBR, CTBP2, CYPB7B1, NOD2 y UBR5. GH también enlaza hacia la regulación del sistema inmune, específicamente hacia la producción del factor de necrosis tumoral donde funcionan los genes APP, BAIAP2, GH, IGF1R, NOD2, SRC, STAT1 y TLR4. De aquí NO2 y TLR4 son el puente hacia otras vías del sistema inmune como lo son activación de citosinas e interleucinas y activación del sistema inmune innato lo realizan; interesantemente en este punto se encuentra de nuevo a LEP quien dirige de nuevo hacia el proceso metabólico de lípidos. Viendo esta vía ontológica funcional desde una perspectiva global, pareciera que el gen GH actúa como un gen maestro que dirige como si fuera el director de una orquesta, la actividad funcional de los demás genes implicados en el desarrollo del ganado lechero. El gen GH está ubicado en el Bta-19 y codifica para la proteína somatotropina. Este gen está asociado PY y MTCAS por el efecto que provoca el SNP rs41923484, que provoca un cambio en el aminoácido 153 de leucina por valina en la somatotropina(23). La somatotropina, también conocida como hormona de crecimiento, participa en múltiples actividades que va desde su efecto en el crecimiento y diferenciación celular de diversos tejidos como es el desarrollo de folículos(38) y de la glándula mamaria(39), así que no es de extrañar que su acción sea clave para que todo el organismo del ganado Holstein funcione de manera adecuada. El observar que la actividad funcional de los 120 genes asociados a 189 SNP dentro de 341 QTL, actúan de manera entrelazada en vías ontológicas que influyen tanto en los procesos de producción, como lo es el desarrollo mamario; así como en vías de reproductivas y de salud, abre la ventana de mejorar la selección para generar animales que vayan poco a poco adquiriendo características tanto productivas como reproductivas.
Agradecimientos
Al CONACYT por el apoyo otorgado en el proyecto no. 216179.
Literatura citada: